
Abstract
In an earlier single-dose escalation study to evaluate the safety and pharmacokinetics of orally administered des-aspartate-angiotensin I (DAA-I) in healthy subjects, the plasma level of DAA-I could not be determined because DAA-I is rapidly degraded in the circulation. The present study investigated the oral bioavailability of DAA-I by measuring the prostaglandin E2 metabolite (PGEM) in the plasma samples of the same trial. PGEM is a stable derivative of PGE2, which has been shown to be a biomarker of DAA-I. The data show that plasma from two of the three subjects who were orally administered the efficacious preclinical dose of 0.70 mg/kg DAA-I exhibited a significant PGEM peak at 5–6 h postdose. Plasma of subjects who were administered 0.08 and 1.5 mg/kg DAA-I, the subefficacious and two-times efficacious dose, respectively, did not exhibit a similar PGEM peak. This observation is concordant with the known in vivo actions of DAA-I, especially its hypoglycemic action where maximum efficacy occurred at a dose of 0.7 mg/kg, and decreased to nil at the two-times efficacious dose. The onset of the PGEM peak at 5–6 h postdose was closed to the 4-h onset of absorption of [C14]DAA-I seen in preclinical rat studies, albeit the absorption kinetics between rodents and humans are not identical. The occurrence of polymorphism of enzymes involved in the formation and degradation of PGE2 is common, and this has been attributed to contributing to the variation in response, onset and peak PGEM observed among the three subjects who were administered the efficacious dose.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The bioavailability of orally administered des-aspartate-angiotensin I (DAA-I) was investigated in human subjects. |
Prostaglandin E2 metabolite (PGEM), a stable metabolite derivative of PGE2 was measured as a biomarker of DAA-I in plasma samples obtained from 15 subjects in a single-dose phase I trial on DAA-I. |
Plasma of two of the three subjects who were administered a preclinical efficacious dose of DAA-I (0.7 mg/kg) showed a significant PGEM peak concentration at 5–6 h postdose. No similar significant PGEM peak was present in the plasma of the other subjects who were administered a subefficacious and a two-time efficacious dose. This observation was in concordance with the known in vivo actions of DAA-I. |
1 Introduction
Des-aspartate-angiotensin I (DAA-I) is an endogenous angiotensin peptide that has been shown to exhibit efficacy in animal models of 11 human pathologies [1,2,3]. DAA-I acts as an agonist on the angiotensin AT1 receptor and releases prostaglandins, which mediate its actions [1, 4]. In cultured human umbilical vein endothelial cells (HUVECs), DAA-I specifically increased the production of prostaglandin E2 (PGE2) and prostaglandin I2 (PGI2) via cyclooxygenase (COX)-1 at sub-nanomolar concentrations, but did not stimulate production of these two prostaglandins via COX-2 [4]. DAA-I was rapidly degraded by circulating enzymes when infused into the systemic circulation of the dog [5, 6] and human [7]. Despite being an endogenous peptide and having a rapid systemic metabolism, DAA-I has been shown to be active when administered orally [1,2,3]. This is probably attributed to its ability to cross Caco-2 cells via passive diffusion [8], indicating that it would cross intestinal epithelial cells, and its activity at concentrations lower than the Km of enzymes [1]. However in a single-dose phase I study, where subjects were orally administered a subefficacious, efficacious, and twice efficacious dose in terms of its preclinical hypoglycaemic activity, we found no significant increase in subjects’ plasma DAA-I concentration over a period of 12 h post-administration [9]. We have advanced various possibilities to explain the absence of DAA-I bioavailability in the phase I study. In a further attempt to obtain physical evidence to show that DAA-I is orally bioavailable, and support its oral efficacy, we measured PGEM, a stable derivative of PGE2 metabolites, as a biomarker in the plasma samples of the single-dose phase I study. The assay was carried out using a commercial ELISA kit, as preliminary experiments showed that the limit of detection for PGE2 and its metabolites, as determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS), was higher than their basal levels in the plasma samples. The results show that two of the three subjects administered the efficacious dose exhibited a plasma PGEM profile, with a maximum at 5–6 h post-administration. No similar profile was seen in the other plasma samples of subjects administered the other two doses of DAA-I.
2 Materials and Methods
2.1 Plasma Samples
Plasma samples were from subjects of a single-dose phase I trial on DAA-I [9]. The samples had been stored frozen at – 80 °C for a period of 18 months, prior to analysis. Three cohorts of six subjects were recruited for the phase I trial and the collected plasma samples were labelled A1–A6, B1–B6, and C1–C6 for Cohorts A, B, and C, respectively. Two subjects in each cohort were administered placebo (sterile water), and the other four subjects from Cohorts A, B and C were administered 0.08, 0.7 and 1.5 mg/kg DAA-I in an aqueous solution, respectively. Volunteers were randomly selected into treatment cohorts. The placebo group plasma samples are denoted with A3, A5, B2, B6, C2 and C5. Due to the use of a plasma sample from each cohort for preliminary analysis in earlier studies, only plasma samples from five subjects in each cohort were available for the present study.
2.2 Plasma Sampling Time
For each subject, plasma samples were taken at 10 different time points, i.e. at time 0 (predosing), and 15 min, 30 min, 1 h, 2 h, 3 h, 4 h, 5 h, 6 h and 12 h postdosing.
2.3 Assay of Prostaglandin E2
PGE2 in the plasma samples was assayed as its stable end metabolite, PGEM, using a Cayman Prostaglandin E Metabolite ELISA Kit (Cat No. 514531; Cayman Chemical, Ann Arbor, MI, USA). The analysis was performed according to the manufacturer’s instructions, as described previously [3]. The Cayman’s assay is based on the competition between a stable PGE metabolite derivative, PGEM, and a PGEM-acetylcholinesterase conjugate (PGEM tracer) for a limited number of PGEM-specific rabbit anti-serum binding sites. PGE2 metabolites in plasma were first converted to PGEM. Briefly, each 100 μL plasma sample was incubated overnight at 37°C with 30 μL of 1 M carbonate buffer. 40 μL of 1 M phosphate buffer and 30 μL of ELISA buffer were added to the plasma mixture. With this treatment, the plasma sample was diluted twice. Eight 100 μL aliquots of PGEM ELISA standard, each aliquot containing an increasing concentration of PGEM, were similarly treated. A 50 μL aliquot of each treated plasma sample was incubated in a well of an assay plate precoated with goat anti-rabbit immunoglobulin G. 50 μL PGEM tracer and 50 μL PGEM ELISA anti-serum were added into each well. After an 18-h incubation at ambient temperature, the plate was rinsed five times with wash buffer. An aliquot of 200 μL freshly prepared Ellman’s reagent was added to each well and the plate was immediately wrapped in aluminum foil and incubated for 90 min in an orbital shaker. Absorbance at 412 nm was read after incubation. The content of PGEM in the eight standard solutions was similarly assayed. Preliminary experiments showed that readings obtained from twice-diluted plasma samples fell within the 20–80% range of the standard curves, and provided the rationale for a two times dilution of the plasma samples.
2.4 Statistical Analysis
One-way analysis of variance was used to test for significance of the increase in PGEM concentrations that occurred between 4.5 and 5.5 h post-administration in plasma samples of subjects B4 and B5 of cohort B. Post hoc analysis was corrected using Tukey’s honest significant difference (HSD) test to correct for the type I error on the pairwise comparisons. Statistical significance was set at p < 0.05.
3 Results
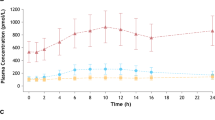
Figure 1 displays the PGEM concentration profile in the 15 plasma samples obtained from subjects who participated in the single-dose phase I trial on DAA-I. The plasma PGEM concentration profile in two of the three samples in cohort B (B4 and B5) had a peak at 5 and 6 h post-administration, respectively. The mean concentration between 4.5 and 5.5 h of the two peak concentrations was significantly higher than the corresponding concentrations in the placebo samples. Additionally, the mean concentration for sample B5 was significantly higher than the corresponding concentrations of the other two samples from DAA-I-treated subjects. The period between 4.5 and 5.5 h post-administration covers the half hour before and half hour after the peak PGEM concentration of sample B4, and the mean plasma PGEM concentrations of cohort B during this period were used for statistical comparison. This period was selected because the peak PGEM concentration of sample B4 was lower than that of sample B5. The PGEM concentration profiles in the plasma samples of cohorts A and C did not exhibit a similar peak concentration.

Plasma PGEM concentrations. A5, B2, B6, C2 and C5 were placebo samples, and samples in the upper, middle and lower plots were from subjects who were administered 0.08, 0.70, and 1.5 mg/kg DAA-I, respectively. * Indicates significantly greater than the corresponding values of B1, B2, B4 and B6 (p < 0.01), # indicates significantly greater than the corresponding values of B2 and B6 (p < 0.01)
4 Discussion
As therapeutics, peptides are recognized for being highly selective, efficacious, and well-tolerated [10]; however, most endogenous peptides are not suitable for use as therapeutics per se because they have a short circulating half-life. Glucagon-like peptide 1, amylin, and somatostatin are examples, and their respective analogues such as exenatide, pramlinitide and octreotide have considerable longer half-life and are used as parental drugs in lieu of the parent peptides. DAA-I, although an endogenous peptide with a similar short circulating half-life, has been shown to be active when administered orally [1,2,3]. Its bioavailability was estimated to be 0.06 in a study where both intravenous and oral DAA-I were shown to produce similar magnitudes of attenuation of cardiac hypertrophy [1]. In preclinical toxicology studies, a [14]C-DAA-I-like peak was detected at 4 h after [14]C-DAA-I was orally administered to rats, indicating that the onset of DAA-I absorption was 4 h post-administration (unpublished preclinical toxicological data). These findings, together with the ability of DAA-I to passage Caco-2 cells via passive diffusion [8], are indications of its oral bioavailability.
The data in Fig. 1 lend further credence to the oral bioavailability of DAA-I. The bioavailability was denoted by the presence of a peak concentration of PGEM, a biomarker of DAA-I, appearing at 5–6 h post-administration in two of the three plasma samples of subjects who were administered the preclinical efficacious dose of 0.7 mg/kg. This observation is in concordance with several known actions of DAA-I. First, DAA-I has been shown to specifically release PGE2 from cultured HUVECs [4]. This enables PGE2 to be a candidate biomarker of DAA-I as the endothelium is the likely tissue of first contact with absorbed DAA-I. HUVECs lack the enzymes that degrade PGE2, but, in systemic circulation, PGE2 is rapidly degraded to its 13,14-dihydro-15-keto metabolites, and in this study was measured as PGEM, the stable derivative of these metabolites [11]. The second commonality is the dose-dependent action of DAA-I, where doses higher than its maximum efficacious dose caused an absence of effect, as shown by the absence of peak plasma concentrations of PGEM in the plasma samples of subjects who were administered a subefficacious (cohort A) and two-times efficacious (cohort C) dose. This dose-related action is also seen in the hypoglycaemic action of DAA-I in the KKAy mouse model of type 2 diabetes, where an efficacy occurred at a dose of 0.7 mg/kg DAA-I; doubling of this dose caused a total loss of the hypoglycaemic effects [12].
The PGEM peak occurred at 5–6 h post-administration in subjects B4 and B5, respectively. This is 1–2 h later than the 4 h absorption onset seen in preclinical studies, and could be due to a variation in absorption kinetics between rodents and humans, or the time taken for the released PGE2 to degrade to its 13,14-dihydro-15-keto metabolites. Variation in response to DAA-I could also have occurred among the DAA-I-treated subjects of cohort B, where subject B1 did not respond, while subject B5 had a more marked response but a later onset than subject B4. The formation of PGE2 involves multiple enzymes [13] and its breakdown to 13,14-dihydro-15-keto metabolites involves both enzymatic and non-enzymatic degradation [11]. The key enzymes involved in the multistep pathway of PGE2 formation and degradation are polymorphic [14, 15], and this could be responsible for the variable responses seen in the three DAA-I-treated subjects of cohort B.
5 Conclusion
The data in the present study are further evidence of the oral bioavailability of DAA-I. Although the bioavailability was determined by measuring PGEM, a stable metabolite derivative of the biomarker PGE2, the observed plasma PGEM profiles were in concordance with the known in vivo actions of DAA-I. This singular matching of plasma kinetics and in vivo actions is a reflection of the specific and unique properties of DAA-I.
References
Sim MK. Des-aspartate-angiotensin I, a novel angiotensin AT1 receptor drug. Eur J Pharmacol. 2015;760:36-1.
Sim MK. The use of des-aspartate-angiotensin I in inflammation related pathologies and diseases. Patent Cooperation Treaty, International Application No. PCT/SG2011/000204, 8 Jun 2011.
Wang H, Sethi G, Loke WK, Sim MK. Des-aspartate-angiotensin I attenuates mortality of mice exposed to gamma radiation via a novel mechanism of action. PLoS One. 2015;10(9):e0138009.
Wen Q, Lee KO, Sim SZ, Xu XG, Sim MK. Des-aspartate-angiotensin I causes specific release of PGE2 and PGI2 in HUVEC via the angiotensin AT1 receptor and biased agonism. Eur J Pharmacol. 2015;768:173–81.
Gayes RP, Szidon JP, Opari S. In vivo and in vitro conversion of des-1-Asp angiotensin I to angiotensin III. Biochem Pharmacol. 1978;27:2871–7.
Sexton JM, Britton SL, Beierwaltes WH, Fiksen-Olsen MJ, Romero JC. Formation of angiotensin III from [des-Asp1] angiotensin I in the mesenteric vasculature. Am J Physiol Heart Circ Physiol. 1979;237:H218–23.
Kono T, Ikeda F, Oseko F, Imura H. Biological activity of des-asp1-angiotensin I in man. J Clin Endocrinol Metab. 1980;50:40–5.
Chua HL, Jois S, Sim MK, Go ML. Transport of angiotensin peptides across the Caco-2 monolayer. Peptides. 2004;25:1327–38.
Lee KO, Khoo CM, Chowbay B, Chan YH, Sim MK. A single dose-escalation study to evaluate the safety and pharmacokinetics of orally administered des-aspartate angiotensin I in healthy subjects. Drugs R D. 2016;16:317–26.
Fosgerau K, Hoffmann T. Peptides therapeutics: current status and future directions. Drug Discov Today. 2015;20:122–8.
Prostaglandin E Metabolite ELISA Kit Item No 51453. Kit Booklet. https://www.caymanchem.com/pdfs/514531.pdf.
Sim MK, Xu XG, Wong YC, Sim SZ, Lee KO. Des-aspartate-angiotensin I exerts hypoglycemic action via glucose transporter-4 translocation in type 2 diabetic KKAy mice and GK rats. Endocrinology. 2007;148:5925–32.
Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119:229–40.
Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, et al. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomark Prev. 2004;13:889–93.
Pereira C, Queirós S, Galaghar A, Sousa H, Pimentel-Nunes P, Brandao C, et al. Genetic variability in key genes in prostaglandin E2 pathway (COX-2, HPGD, ABCC4 and SLCO2A1) and their involvement in colorectal cancer development. PLoS One. 2014;9(4):e92000.
Acknowledgements
The study was supported by Grant R-184-000-237-511 from the National Medical Research Council, Singapore
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kok-Onn Lee, Edmund Feng Tian, Martin Hui Cai, Hong Wang, Yiong-Huak Chan and Meng-Kwoon Sim have no conflicts of interest with regard to financial sponsorship or governmental guidelines on the publication of this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lee, KO., Tian, E.F., Cai, M.H. et al. Bioavailability of Orally Administered Des-Aspartate-Angiotensin I in Human Subjects. Drugs R D 18, 51–54 (2018). https://doi.org/10.1007/s40268-017-0218-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0218-4