
Abstract
Fibrosis and its end-stage cirrhosis are the result of chronic inflammatory liver disease. Historically considered irreversible, fibrosis has recently been demonstrated to be reversible. In this respect, the liver is a germane model to review extracellular matrix (ECM) remodelling and the fibrotic process. Upon injury hepatic stellate cells (HSCs) become activated and produce ECM components, including fibrillar collagens. The balance between ECM deposition and ECM degradation is skewed towards deposition by an increase expression of Tissue Inhibitors of Matrix metalloproteinases (TIMPs) versus Matrix Metalloproteinases (MMPs). In experimental models of spontaneous fibrosis resolution, liver TIMP levels decrease associated with HSC apoptosis or phenotypic reversion to quiescence; macrophages play a pivotal role in fibrolytic activity by increasing the expression of MMPs, thereby facilitating ECM degradation. Further investigations on the control of ECM deposition and degradation will likely facilitate the design of effective therapies aiming at resolving fibrosis by enhancing ECM degradation.
Similar content being viewed by others

Introduction
Fibrosis is a highly conserved protective response to repetitive or chronic tissue injury. The interaction of multiple pathways, molecules and systems determines whether fibrosis is self-limiting or whether it is uncontrolled and excessive [1••]. Given the number of organs that can be affected by fibrosis it would be impossible to describe the process exhaustively in each of them. Therefore, this review will focus on mechanisms underlying the biology of the extracellular matrix in liver fibrosis, a process that we and other have shown in rodents and humans to be bidirectional. As many fibrogenic pathways are conserved across tissues, recent findings in the liver may partially be extended to studies of fibrosis in the lungs, the kidneys, the heart and other organs. Indeed, the idea that there are conserved core and regulatory pathways in fibrosis has been suggested to help identify the most promising generic anti-fibrotic targets [2]. Further the liver constitutes a useful model to define the interplay between several players of the mammalian wound-healing response including the epithelial, inflammatory, myofibroblast and extracellular matrix (ECM) components [1••]. It has been recently shown that the development of liver fibrosis is a dynamic and potentially bidirectional process: both in human trials and in rodent models in which the hepatic injurious stimulus is successfully removed, spontaneous resolution of scarring is reported [3]. In this context, the physiology of ECM deposition and remodelling is of particular interest. In the case of reversible fibrosis, an uneven balance between deposition and degradation of ECM exists, while the end-stage of liver fibrosis, cirrhosis, which is incompletely reversible is characterised by the development of paucicellular scars enriched in extensively cross-linked matrix components, such as fibrillar collagen and elastin [3], although whether these factors or others determine irreversibility has yet to be completely defined.
Key Concepts About Liver Fibrosis Establishment and Resolution
Germane to understanding of the molecular mechanisms underlying liver fibrosis establishment and resolution is the knowledge of the specific histological organisation of the organ and of the major cellular players involved.
The liver is divided into lobules in which hepatocytes are lined by sinusoidal capillaries that converge towards a central efferent vein. Liver lobules are hexagonal and at each corner of the hexagon a portal triad of vessels is located. The portal triad consists of a portal vein, a hepatic artery and a bile duct. Sinusoids are liver-specific structures that thanks to a peculiar anatomy favour the metabolite exchange to and from the liver parenchyma (or hepatocytes). Sinusoids are constituted by capillaries with fenestrated endothelial cells, behind these, in the space of Disse adjacent to the hepatocyte palisade lie hepatic stellate cells, resident macrophages (also named Kupffer cells) and large granular lymphocytes. The liver as a whole organ has a dual blood supply, via the portal vein and the hepatic artery. The former brings into the liver the venous blood from the intestines, pancreas and spleen, while the latter supplies the liver with oxygen. The portal triad is instrumental to microcirculation into lobules since the blood flows from the portal triad through a sinusoidal capillary to a central efferent vein located in the middle of the lobule [4].
The major metabolic functions of the liver are carried out by the hepatocytes which constitute the major parenchymal mass and represent almost 60 % of the total liver cells and about 80 % of the volume of the organ. Hepatocytes are highly polarized epithelial cells: the basolateral membrane facilitates the transfer of materials to and from the fenestrated capillaries; it is exposed to across the space of Disse, while tight junctions formed between hepatocytes create a canaliculus surrounding each hepatocyte. The canaliculi deliver bile salts produced in hepatocytes to bile ducts at the portal triad. A specialised epithelial cell, the biliary epithelial cell or cholangiocyte forms the bile ducts [4].
The hepatic stellate cell (HSC) has been described to have a pivotal effector role in fibrogenesis. In steady-state condition, HSC resides in the space of Disse and is a fat and retinoid ester storage cell. During acute and chronic injury, the quiescent HSC turns to an “activated” myofibroblast-like phenotype after its retinoid and lipid droplets are shed. ECM production is one of the distinctive features of activated HSC; particularly, they express and secrete fibrillar collagen and contribute to fibrotic matrix deposition. Activated HSCs also express tissue inhibitors of metalloproteinases (TIMPs) as well as chemotactic and vasoactive factors. Among these TIMP-1 is expressed at a high level and inhibits a wide range of matrix metalloproteinases (MMPs), preventing them digesting ECM components hence favouring scar accumulation [5••]. To activate and perpetuate activation of HSCs, cell–matrix interactions may be important. Studies in tissue culture indicate that loss of the normal liver matrix and its replacement with a matrix rich in collagen I may promote HSC activation [6]. Also hepatocyte function can be perturbed by altered cell–matrix interactions (see below). At final stages of liver fibrosis, the architecture is completely disrupted by dense condensations of collagens form, which cross-link together the vascular structures and as a consequence the common clinical symptoms of advanced fibrosis/cirrhosis appear [7].
The extracellular matrix in the healthy liver is composed of several classes of macromolecules which include collagens (types I, III, IV, V and VI) and the non- collagenous glycoproteins such as laminin and fibronectin amongst others and proteoglycans [8]. The normal liver sinusoid is lined by a non-electron dense basement membrane matrix composed by laminin and type IV collagen. During the fibrosis development, the healthy matrix undergoes dramatic changes. Interstitial collagens, particularly collagens I and III, replace the physiological ECM components in the space of Disse. In the early phases of fibrosis, the accumulation of fibrillar ECM is associated with a loss of endothelial fenestrae and a morphological modification of the hepatocytes that lose their microvilli. This phenomenon, known as capillarisation of the sinusoids, is the result of changes in cell–matrix interactions [8–10]. Progressively areas of hepatocyte regeneration form spheres which further distort the liver architecture. Concomitantly vascular structures are linked and angiogenesis may be observed in dense areas of scarring [8, 10]. Experimental models show a 4- to 7-fold increase in the content of collagen and glycosaminoglycans in the cirrhotic liver as compared with healthy control livers. Together with fibrillar collagens, there is an increase in non-collagenous components such as laminin and proteoglycans [8], with cross-linking of matrix also documented [10]. In advanced fibrosis, termed cirrhosis, a dense ECM containing elastin is deposed alongside the fibrillar collagens and the non-collagenous components described. Indeed, elastin is used as a pathological marker for the presence of chronic fibrosis. Impairment of hepatic function and risk of hepatocellular cancer development are linked with the profound changes in matrix composition as described above [11]. The dramatic architectural changes lead to the development of the clinical signs and symptoms of liver cirrhosis such as portal hypertension, a leading cause of mortality among cirrhotic patients [8].
It is important to underline that fibrosis is not an irreversible process: evidence suggests that even as fibrosis progresses matrix degradation is possible. Key to fibrosis remodelling is the action of MMPs. MMPs are a family of zinc and calcium dependent endopeptidases which are produced by connective tissue cells and inflammatory cells and have a range of activity against the major constituents of ECM including fibrillar and non-fibrillar collagens and elastin [7]. Distinct MMPs show specificity for distinct substrates, with some characterised by a more promiscuous range of targets, while others are more substrate specific. Several cell types express MMPs in the liver: hepatocytes, hepatic stellate cells, Kupffer cells and neutrophils and recruited hepatic macrophages [7, 12]. MMPs are classified on the basis of their substrate specificity. For a complete classification of MMPs, please refer to Table 1. Collagenases are one of the sub-families in which MMPs may be grouped. They play a pivotal role in ECM remodelling since they are endowed with the ability to digest the native helix of fibrillar collagens thereby generating a gelatin susceptible to degradation by other MMPs [13]. Distinct collagenases are expressed by distinct cell types at specific stages of liver fibrosis. For example neutrophils and macrophages, both present in the liver during the early inflammatory phases of fibrosis establishment express the neutrophil collagenase, MMP-8. Inflammatory cells in the human liver express the interstitial collagenase or MMP-1, and its murine counterpart MMP-13 is produced by stellate cells and macrophages [5••, 14] in the rodent. Specific cell types show a distinct regulation of the same MMP. MMP-13 is a hallmark of early-activated HSC, while they lose its expression when they become fully activated. Macrophages instead express the molecule quite constantly regardless of the stage or stimulus for activation in models of liver injury. Interstitial collagenases have traditionally been considered active only against fibrillar collagen, but there are reports that gelatinase A (MMP-2) may have a similar function [5••]. The stromelysins have a promiscuous substrate profile and activity against collagens, laminin and fibronectin [15]. Inflammatory cells and HSCs express stromelysins during liver fibrosis. The expression of the stromelysin(MMP-3) is linked to early activation of the HSC and it has been postulated that MMP-3 expression is responsible for basement membrane degradation in early stage of fibrogenesis. Another sub-family characterised by a wide range of substrates is the one of gelatinases. Gelatinase A (MMP-2) has activity against gelatins, collagens IV, V, VI, VII, X and XI and also some elastase and collagenase activity. Gelatinase B (MMP-9) shares a similar substrate profile but with the exception of the collagenase activity. Kupffer cells and inflammatory macrophages express MMP-9; MMP-2 has been described in activated HSCs [5••]. Metalloelastase (MMP-12) is an MMP with potent elastin-degrading activity. Using experimental models in genetically modified mice, we have recently reported its expression of MMP12 by hepatic macrophages and described the degradation of elastin in models of hepatic fibrosis is dependent on this enzyme [16].
The imbalance between fibrosis deposition and degradation in favour of the former determines the transition from a physiological to a pathological ECM composition in fibrotic liver. Tissue Inhibitors of MMPs (TIMPs) are pivotal player in the process, since they control the extracellular activity of MMPs. At present four TIMPs have been described [17]. In two rat models of liver injury in rodents, CCl4 injection and bile duct ligation (discussed below), an up-regulation of TIMP-1 and TIMP-2 has been observed [18]. HSCs are the main producers of TIMP-1 and TIMP-2 [7] and various human liver diseases are associated with their overexpression [19]. Although TIMP-1 production could be clearly attributed to activated HSCs [20] in early stages of liver injury Kupffer cells and hepatocytes may also contribute to its production. The reported pattern of expression of TIMP-1 has been confirmed in animal models of chronic liver injury [20–22]. TIMP-1 protects newly synthesized collagen from degradation by MMPs through the binding and inhibition of activated collagenases. TIMP-1 is also able to prevent the activation of pro-MMPs [17]. And through the inhibition of MMPs, TIMP-1 is also able to prevent the induction of HSC apoptosis, a key step in fibrosis resolution phase [23] (see below). Overexpression of TIMP-1 prevents matrix degradation in progressive injury and spontaneous resolution [24]. TIMP-1 is also significantly associated with fibrogenesis in other organs such as the lung, kidney, and pancreas; for this, it has been proposed as a central molecule in the general process of fibrosis establishment and resolution [17]. TIMP-2 mRNA expression has been described in rat liver myofibroblasts, activated HSCs and Kupffer cells but not in healthy hepatocytes [25]. Acute toxic liver injury induced in rats by a single injection of CCl4 is associated with TIMP-2 transcript expression peaking transiently after 48 h. In models of chronic toxic liver based on CCl4 administration for 8 weeks, TIMP-2 mRNA does not increase [17], while in bile ductal ligation (BDL)-induced fibrosis TIMP-2 mRNA is elevated after day 10, as collagen becomes laid down [21]. The requirement for TIMP-2 to activate MMP-2 has been demonstrated using TIMP-2 KO mice [26].
Models to Study Liver Fibrosis
Our understanding of the pathogenesis of liver fibrosis has been facilitated by the use of complimentary experimental models. Models of liver fibrosis can be assigned to three broad groups, each of which has specific advantages and disadvantages [27].
The first group includes cell culture models, instrumental to the fine study of cell behaviour and the effect of specific mediators. By extracting highly pure primary cells from normal or experimentally injured livers, sophisticated cell biology studies are possible in vitro. However, cultured cells cannot recapitulate the complexity of events that occur in vivo, deriving from the interplay of resident and incoming cells in the liver microenvironment [27].
The second group includes human tissues taken at biopsy or following hepatic resection. Investigating human samples is fundamental to confirm findings in cell cultures and animal models. Advanced molecular techniques have enhanced significantly the amount. A major drawback of the use of human samples is that they tend to be representative of only one time point, since very reasonable ethical concerns prevent multiple sampling over time in the same patient. Moreover, data obtained at surgery or through biopsy are generally, but not exclusively, representative of fairly advanced stages of disease, and miss the very early events in fibrosis [27]. Nevertheless, a good example of the usefulness of those models is represented by large trials of antiviral therapies in chronic hepatitis B and C that have demonstrated remodelling of fibrosis and architectural restoration [3, 10, 27–29].
To address and resolve the issues raised by the use of the first two types of model, as well as to describe a potentially dynamic process, a third type of model has proven important: Experimental animal models of liver fibrosis [27]. The biomarkers for liver insults are similar, and sometimes even identical, in humans and animals and include transaminases and other blood tests, biopsy and non-invasive imaging techniques [30]. Several approaches to induction of fibrosis have been described. Of these, CCl4 intoxication in rats and mice is probably the most widely studied. In addition, the CCl4 model is best characterised in terms of the histological, biochemical, cellular, and molecular changes associated with the development of fibrosis. CCl4 can be given intraperitoneally or by oral gavage; it induces necrosis and hepatocyte apoptosis with associated hepatic stellate cell activation and tissue fibrosis. CCl4 can be administered once to study the impact of an acute injury to liver mechanisms of tissue remodelling. Repetitive injections of CCl4 can be used to induce bridging hepatic fibrosis (4 weeks of twice-weekly dosing), cirrhosis (8 weeks of twice-weekly dosing) and advanced micronodular cirrhosis (12 weeks of twice-weekly dosing). For each of these models, spontaneous recovery from fibrosis can be studied after cessation of CCl4 administration. Mechanistic studies using gene knockout and transgenic animals can also be performed using CCl4 [31].
Cholestatic liver injury is one of the major causes of liver fibrosis and cirrhosis in patients with acute or chronic liver disease. Damage to the biliary epithelium and bile duct injury may lead to end-stage liver disease, liver failure and need for organ transplantation, and it is a life-threatening condition [30]. Bile duct ligation (BDL) is a classic experimental model for secondary biliary fibrosis [32••]. BDL stimulates the proliferation of biliary epithelial cells, resulting in proliferating bile ductules, cholestasis, portal inflammation and fibrosis, and ultimately leads to liver failure [33]. BDL has been used also to study reversibility of liver fibrosis, since spontaneous resolution of biliary fibrosis has been observed in rats following biliojejunal anastomosis [33].
Gatekeepers of Fibrosis Resolution Process
Deciphering Spontaneous Resolution of Fibrosis
Using the above models, several distinct patterns of TIMP and MMP expressions have been characterised during spontaneous resolution of fibrosis. The molecular changes observed during the phase of fibrosis resolution include a reduction in the production of fibrillar collagen and a in expression of TIMPs, resulting in a change in MMP activity [5••]. Fibrillar collagen degradation results from an overall increase in liver collagenase activity; collagenases such as MMP-1, -8, -13 and -14 are likely to be responsible for key events in the initial degradation of ECM. The initial cleavage of collagen type I is in fact a key step in the process of fibrosis resolution as demonstrated in mice expressing a mutated cleavage-resistant collagen type I that cannot solve fibrosis [34]. Constant MMP-13 expression has been observed using ribonuclease protection assay during a 28-day spontaneous recovery from CCl4-induced liver fibrosis [35]. In a double-transgenic mouse model with inducible liver-specific expression of TGF-b after switching off TGF-b expression, a continuous MMP-13 expression coinciding with reduced TIMP-1 expression has been observed during fibrolysis, probably as a consequence of synthesis by Kupffer cells [36]. MMP-2 has been at first considered important for fibrosis establishment being expressed by activated HSC [37]. However, expression of MMP-2 during fibrosis resolution allows immediate metalloproteinase reactivity as soon as TIMP-1 levels declined [38]. Moreover, it has been shown that pro-MMP-2 activation occurs following HSC apoptosis (a key cellular attribute of fibrosis resolution, see below) [39]. MMP-2 and MMP-14 may both exhibit gelatinolytic as well as interstitial collagenolytic properties [40, 41]. Models of CCl4 intoxication show increased levels of expression of MMP-2 during spontaneous recovery. Results have been confirmed at protein level using zymography on total liver extracts [38, 42]. MMP-2 may also important by providing an increased gelatinolytic activity in liver fibrosis resolution contributing to the more complete degradation of partially denatured fibrillar collagens [25, 35, 43]. In several experimental models, up-regulation of MMP-9, another gelatinase, has been demonstrated at mRNA level and protein level during resolution. MMP-9 does not exhibit clear collagenase activity against fibrillar collagen I but is sharply increased at the onset of fibrolysis MMP-9; therefore, an indirect involvement of MMP-9 in fibrolysis has been hypothesised [17]. Activated HSCs rely on expression of avb3 integrin to provide themselves with pro-survival signals, since avb3 mediates cell–matrix (type I collagen) interactions [34].
To define the specific contribution of each cell types in the expression of MMPs and TIMPs, gene array and molecular studies are useful techniques, particularly if combined with topographic location analyses such as ISH and IHC. However to define mechanistically the contribution to fibrosis establishment and resolution of individual cell types and mediators, genetically modified mice that allow cell tracking or deletion of specific cell lineages or genes have proven invaluable. One of the key defining features of spontaneous resolution of liver fibrosis is apoptosis of myofibroblast-like hepatic stellate cells [44]. Using a model of CCl4 injection for 4 weeks in rats, it has been demonstrated that apoptosis of activated HSC is a key factor in regression of liver fibrosis. The same has been demonstrated using the BDL model in rats. Moreover it is clear that increased apoptosis of activated HSC correlates with decreased expression of TIMPs, thus tipping the balance of MMPs/TIMPs towards ECM net degradation, a key event in determining fibrosis resolution [35, 44, 45]. Activated HSC are susceptible to apoptosis, and pharmacological therapies targeting activated HSCs to undergo apoptosis have proven to be effective in vivo in reducing fibrosis in experimental models [46, 47]. In the control of activated stellate cell survival, the ECM itself may play a pivotal regulatory role. ECM is regarded as a key mediator of cell survival and proliferation. It has been demonstrated that intact collagen-1 promotes persistence of activated HSC in vivo. Additional studies in mice carrying a mutated form of collagen-1 that confers resistance to the action of collagenase demonstrate that degradation of collagen-1 is critical to induction of HSC apoptosis during recovery from liver fibrosis [34, 44, 48]. Other cell types are involved in the control of HSC activation and apoptosis. It has been demonstrated in vitro that KCs, when stimulated with LPS, induce apoptosis of HSCs via caspase 9 but not caspase 3 or 8 activation [49•].
Recently it has been postulated that HSCs apoptosis might be one of at least two pathways leading to fibrosis regression in damaged liver. In an elegant study using a combination of quantitative polymerase chain reaction at single cell level and a transgenic mouse model that expresses a tamoxifen-inducible CreER under the control of vimentin promoter, it has been shown that activated HSCs may undergo a phonotypic reversion to a quiescent state, thereby contributing to resolution and architectural restoration during spontaneous recovery from experimental fibrosis [50]. In a parallel study using a model of CCl4 intoxication, others have demonstrated that half of the myofibroblasts escape apoptosis during regression of liver fibrosis, down-regulate fibrogenic genes and acquire a phenotype similar to quiescent HSCs. However, differently from unstimulated quiescent HSCs, the reverted population retains the ability to more rapidly reactivate into myofibroblasts in response to fibrogenic stimuli. Inactivation of HSCs has been described to be associated with up-regulation of the anti-apoptotic genes Hspa1a/b, which participate in the survival of HSCs both in vitro and in vivo [51••].
Either phenotypic conversion or apoptosis of HSC would be expected to be associated with a down-regulation of TIMPs in the liver. Consistently this has been documented in models of fibrosis recovery. However it is difficult to postulate that a cell population declining as a result of either process is the major source of MMPs, that promote ECM degradation and fibrosis regression. Further, studies in which activated HSC/myofibroblasts have been deleted via apoptosis show enhanced fibrosis remodelling, suggesting that HSC are not necessary for resolution. These observations have led to a focus on macrophages, and latterly dendritic cells, as potential sources of MMPs critical to fibrosis resolution [1••, 3, 5••, 6].
Macrophages Plasticity in Liver Fibrosis
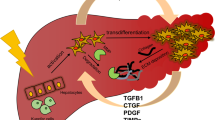
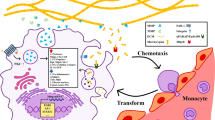
Cells of the monocyte/macrophage lineage are key players of the innate immune response following tissue damage (Fig. 1). A number of experimental studies implicate macrophages in promoting hepatic inflammation and fibrosis [6, 52••]. In the injured liver infiltrating macrophages are located in close proximity to areas of fibrosis and activated hepatic myofibroblasts and produce factors such as TGF-b, IL-1b, PDGF and CCL2 which in turn enhance the pro-fibrogenic gene expression of the activated HSC. Particularly, macrophage-derived moieties in the pro-fibrogenic phase promote activation, proliferation, chemotaxis and survival of HSCs [53].

Relationship between macrophages and ECM biology during fibrosis establishment and fibrosis resolution. During fibrogenesis, inflammatory monocytes are recruited to the inflamed liver, giving rise to the pro-fibrotic macrophage population. Pro-fibrotic macrophages express inflammatory mediators such as lI-1b, TGF-b and PDGF, which promote activation of HSCs. Activated HSCs synthesize the ECM and TIMP-1. Both TIMP-1 and ECM interactions promote persistence of the activated myofibroblast phenotype. During fibrosis resolution, there is a change in macrophage phenotype. It is still not clear if the change arises from a phenotypic switch of pro-fibrotic macrophages or a separate recruitment of monocytes. Pro-resolution macrophages express MMPs that promote ECM degradation. Pro-resolution macrophages express mediators that induce activated HSCs apoptosis, leading to a reduction in ECM synthesis, loss of TIMP-1 expression and enhanced MMP activity
CCL2 is one of the main chemoattractants for pro-inflammatory monocytes in the damaged area of virtually any organ. Functional studies using inhibitor of CCL2 or CCL2 knockout animals have shown a reduction of hepatic macrophage infiltration and of fibrogenesis following chronic injury. We have recently shown that macrophages may play a dual role in liver fibrosis establishment and resolution [54, 55]. An elegant paper from Karlmark and colleagues identified a subset of hepatic macrophages expressing Gr-1 at high levels on their surface and derived from recruitment of inflammatory monocytes via CCL2/CCR2 interaction as the main population controlling the pro-fibrogenic phase [56]. However, if macrophage depletion is achieved experimentally after recruitment to the damaged liver, then fibrosis resolution is impaired. Put another way, macrophage depletion has completely opposite effects if performed in the phases of fibrosis establishment and fibrosis resolution [54]. Ramachandran et al. have defined the population of macrophages acting in the phase of fibrosis resolution as restorative macrophages (RMφs). To identify the hitherto elusive RMφ the authors have used the model of liver damage induced by CCl4 injection. The RMφ has been identified as a CD11Bhi F4/80int Ly-6Clo macrophage subset derived from infiltrating inflammatory macrophages and representing the principle MMP-expressing subset. Depletion of the RMφ population results in a failure of scar remodelling. Further experiments have shown that the RMφs are derived from recruited Ly-6Chi monocytes, a common origin with pro-fibrotic Ly-6Chi macrophages. This has been proposed as a evidence for a phenotypic switch in vivo that converts pro-fibrotic macrophages into macrophages endowed with pro-resolution properties (RMφs). Microarray profiling of the Ly-6Clo subset (RMφ), compared with Ly-6Chi subset (pro-fibrotic macrophages), has shown a phenotype outside the conventional M1/M2 classification. In fact, RMφs display increased expression of MMPs (including Mmp9, Mmp12), growth factors (e.g. Igf1) and phagocytosis-related genes (e.g. Gpnmb). Confocal microscopy confirmed the post-phagocytic nature of the RMφ. Furthermore, the RMφ phenotype has been recapitulated in vitro by the phagocytosis of cellular debris. Notably if a phagocytic behaviour is induced in vivo by the administration of liposomes, the number of RMφs increases and this in turn accelerates fibrosis resolution [57••]. Scar-associated macrophages are also regulated by Vascular Endothelial Growth Factor [58] in repairing liver. During fibrosis resolution, blockade of VEGF causes an impaired liver sinusoidal permeability with a consequent decrease in monocyte migration [59].
Conclusions
Over the last 30 years our view of liver fibrosis has changed dramatically. Whereas before liver fibrosis was considered irreversible, there is now cogent evidence for significant fibrosis reversibility in both human and rodent models. In turn, these models have provided with the opportunity to dissect out the key players determining reduction of fibrosis. It is to be anticipated that this knowledge will lead to the development of specific or targeted anti-fibrotic (or, more accurately, pro-resolution) therapies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Pellicoro A et al (2014) Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 14(3): 181–94. This review is an excellent point of reference as sum-up of the intimate interplay between inflammation and liver fibrosis establishment and resolution
Mehal WZ, Iredale J, Friedman SL (2011) Scraping fibrosis: expressway to the core of fibrosis. Nat Med 17(5):552–553
Pellicoro A, Ramachandran P, Iredale JP (2012) Reversibility of liver fibrosis. Fibrogenesis Tissue Repair 5(Suppl 1 Proceedings of Fibroproliferative disorders: from biochemical analysis to targeted therapiesPetro E Petrides and David Brenner): S26
Tanaka M et al (2011) Liver stem/progenitor cells: their characteristics and regulatory mechanisms. J Biochem 149(3):231–239
•• Iredale JP, Thompson A, Henderson NC (2013) Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim Biophys Acta 1832(7): 876–83. In this review the authors give a complete and comprehensive review of the cellular and molecular player involved in liver fibrosis establishment and resolution. Of particular importance for the neat description of the reversible nature of the fibrotic process
Ramachandran P, Iredale JP (2012) Liver fibrosis: a bidirectional model of fibrogenesis and resolution. QJM 105(9):813–817
Iredale JP (1997) Tissue inhibitors of metalloproteinases in liver fibrosis. Int J Biochem Cell Biol 29(1):43–54
Friedman SL (1993) Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med 328(25):1828–1835
Friedman SL et al (1985) Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci USA 82(24):8681–8685
Issa R et al (2004) Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 126(7):1795–1808
Zhang DY, Friedman SL (2012) Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 56(2):769–775
Consolo M et al (2009) Matrix metalloproteinases and their inhibitors as markers of inflammation and fibrosis in chronic liver disease (Review). Int J Mol Med 24(2):143–152
Siller-Lopez F et al (2004) Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology 126(4):1122–1133 discussion 949
Fallowfield JA et al (2007) Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 178(8):5288–5295
Docherty AJ et al (1992) The matrix metalloproteinases and their natural inhibitors: prospects for treating degenerative tissue diseases. Trends Biotechnol 10(6):200–207
Pellicoro A et al (2012) Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 55(6):1965–1975
Hemmann S et al (2007) Expression of MMPs and TIMPs in liver fibrosis—a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol 46(5):955–975
Roeb E et al (1997) TIMP expression in toxic and cholestatic liver injury in rat. J Hepatol 27(3):535–544
Benyon RC et al (1996) Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology 110(3):821–831
Iredale JP et al (1996) Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology 24(1):176–184
Kossakowska AE et al (1998) Altered balance between matrix metalloproteinases and their inhibitors in experimental biliary fibrosis. Am J Pathol 153(6):1895–1902
Bergheim I et al (2006) Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther 316(2):592–600
Mohammed FF et al (2005) Metalloproteinase inhibitor TIMP-1 affects hepatocyte cell cycle via HGF activation in murine liver regeneration. Hepatology 41(4):857–867
Yoshiji H et al (2002) Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology 36(4 Pt 1):850–860
Knittel T et al (1999) Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol 30(1):48–60
Wang Z, Juttermann R, Soloway PD (2000) TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem 275(34):26411–26415
Iredale JP (2007) Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 117(3):539–548
Iredale JP (2001) Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis 21(3):427–436
Ramachandran P, Iredale JP (2009) Reversibility of liver fibrosis. Ann Hepatol 8(4):283–291
Liedtke C et al (2013) Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 6(1):19
Constandinou C, Henderson N, Iredale JP (2005) Modeling liver fibrosis in rodents. Methods Mol Med 117:237–250
•• Liu Y et al (2013) Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol 304(5): G449-68. This is a seminal point of reference regarding rodent models of chronic liver injuries. It offers a systematic review of distinct models clearly stating advantages and disadvantages of the use of each of them
Kountouras J, Billing BH, Scheuer PJ (1984) Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol 65(3):305–311
Issa R et al (2003) Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J 17(1):47–49
Iredale JP et al (1998) Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 102(3):538–549
Arendt E et al (2005) Enhanced matrix degradation after withdrawal of TGF-beta1 triggers hepatocytes from apoptosis to proliferation and regeneration. Cell Prolif 38(5):287–299
Okazaki I et al (2000) Molecular mechanism of the reversibility of hepatic fibrosis: with special reference to the role of matrix metalloproteinases. J Gastroenterol Hepatol 15(Suppl):D26–D32
Zhou X et al (2004) Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem 279(23):23996–24006
Preaux AM et al (2002) Apoptosis of human hepatic myofibroblasts promotes activation of matrix metalloproteinase-2. Hepatology 36(3):615–622
Aimes RT, Quigley JP (1995) Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J Biol Chem 270(11):5872–5876
Ohuchi E et al (1997) Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem 272(4):2446–2451
Takahara T et al (1995) Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 21(3):787–795
Watanabe T et al (2001) Dynamic change of cells expressing MMP-2 mRNA and MT1-MMP mRNA in the recovery from liver fibrosis in the rat. J Hepatol 35(4):465–473
Elsharkawy AM, Oakley F, Mann DA (2005) The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 10(5):927–939
Kisseleva T, Brenner DA (2006) Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 21(Suppl 3):S84–S87
Salvesen GS, Abrams JM (2004) Caspase activation - stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene 23(16):2774–2784
Wright MC et al (2001) Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology 121(3):685–698
Issa R et al (2001) Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 48(4):548–557
• Fischer R et al (2002) Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology 123(3): 845–61. This is a seminal paper for the understanding of the role of macrophages in liver fibrosis. It offers for the first time a formal proof of the plasticity of macrophages and their impact on development and resolution of liver fibrosis
Troeger JS et al (2012) Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143(4): 1073–83 e22
•• Kisseleva T et al (2012) Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA 109(24): 9448–53. This paper is of pivotal importance in defining a possible new pathway of reduction of HSCs activity in the phase of fibrosis resolution: beside apoptosis, deactivation to a quiescent-like state of HSCs could take place in the regenerating liver
•• Thomas JA et al (2011) Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 53(6): 2003–15. See comment on ref. 57
Wynn TA, Barron L (2010) Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30(3):245–257
Duffield JS et al (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115(1):56–65
Baeck C et al (2014) Pharmacological inhibition of the chemokine C–C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 59(3):1060–1072
Karlmark KR et al (2009) Hepatic recruitment of the inflammatory Gr1 + monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 50(1):261–274
•• Ramachandran P et al (2012) Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA 109(46): E3186–95. The two papers are germane to the full understanding of the role of macrophages in liver fibrosis. they offer a view of the phenotype that macrophages acquire in the process and how they could be exploited as cellular therapy to solve fibrosis. Ref. 54 offers for the first time a mechanistic and molecular description of the hiterto elusive resotrative macrophages
Ehling J et al (2014) CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut
Yang L et al (2014) Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology 146(5): 1339–50 e1
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Campana, L., Iredale, J.P. Extracellular Matrix Metabolism and Fibrotic Disease. Curr Pathobiol Rep 2, 217–224 (2014). https://doi.org/10.1007/s40139-014-0058-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-014-0058-7