
Abstract
In the present study, different process intensification options in batch mode are considered for a class of reactions, and are evaluated with respect to enhancement in conversion and product purity considering methyl acetate formation reaction as an experimental case study. The options explored include operation in different batch operating units and use of different molar ratios of reactants. Integrated reaction and separation is considered in two ways, namely by simple reactive batch distillation, and by multi-stage reactive batch distillation with partial reflux. It is observed that conversions beyond equilibrium conversion are achieved by process intensification, and relatively purer products are obtained compared to the base case. The purity and conversions in the multi-stage column are found to be higher than with simple distillation. Increasing the molar ratio of methanol in the reaction feed mixture is found to yield high conversions at the expense of purity of methyl acetate in the product. The present study considers a typical case of an esterification reaction with azeotropic mixtures, and the analysis is applicable to a class of reactions satisfying the criteria mentioned.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the frame of globalization and sustainability, process intensification is a path to the future chemical and process engineering. Process intensification converts “molecules into money” which is based on the premise that the chemical engineering drives today’s economy development and is fundamental to wealth creation [1]. Process intensification (PI) has been defined as “Any chemical engineering development that leads to a substantially smaller, cleaner and more energy-efficient technology” [2]. Process intensification can be achieved by using new equipment [3], or by employing different methods in previously well-known equipment. Further, the new equipment is classified as reactors and equipment for non-reactive operations, whereas the methods have been viewed as multifunctional reactors, hybrid separations [4], alternate renewable raw materials [5], alternate energy sources [6] and other methods. The main focus while exploring PI options for a given process has been continuous operation, since a shift from batch to continuous operation automatically leads to smaller and more energy-efficient operation. In this context, extensive literature has been reported over the past decade, and reactive separations in general and reactive distillation in particular have occupied one of the top slots among the PI studies reported. Harmsen [7] provided a full-scale review of commercial applications, research, scale-up, design and operation of reactive distillation, showing it to be the front runner of industrial process intensification, and stated that reactive distillation has been implemented in more than 150 commercial operations of petrochemical and chemical industry. Sharma and Mahajani [8] and Hiwale et al. [9] have provided exhaustive reviews on the industrial applications of reactive distillation covering several classes of reactions including esterification, trans-esterification, hydrolysis, etherification, hydrogenation, hydrodesulphurization, dehydrogenation, alkylation, metathesis, hydration, dehydration, carbonylation, polymerization, chlorination, amination, synthesis of carbonates, chiral separation and acetalization along with industrial examples for each class of reaction, and have pointed out some inherent limitations which can be overcome through proper choice of parameters. Taylor and Krishna [10] have provided a comprehensive review on different modeling approaches applicable for reactive distillation.
Although batch process operation may not be classified as process intensification by default, the chemical industry has re-oriented from large-volume low-value production in continuous processes towards high-value low-volume production in batch processes due to the demand for a large number of specialty chemicals. Batch operations with or without reaction have become attractive to the chemical, food and pharmaceutical industry for products with high added value and more operational flexibility. Therefore, it is necessary to explore PI options in batch operation also to derive the maximum benefits within the available constraints. Reactive batch distillation processes hold considerable promise in the current competitive environment as they have the combined benefits of integrated process operation and multi-component separation in a single unit. The synergistic effect of the combined operation has the potential to increase conversion, improve selectivity, reduce the capital investment and provide additional flexibility. Bollyn and Wright [11] and Gadewar et al. [12] have presented a method to estimate the advantage of using batch reactive distillation over conventional technology in terms of yield and selectivity.
In the present study, it is attempted to explore the possible options for process intensification in batch mode for a class of reactions, considering homogeneously catalyzed methyl acetate production as an experimental case study. The objective is to achieve high conversion as well as high-purity methyl acetate product in batch operation while using a homogeneous catalyst. The details regarding PI approaches considered will be presented in the next section, followed by a case study of methyl acetate production with different PI options explored.
Process intensification schemes in batch mode
Process intensification studies have already been well-established in continuous operation, especially using reactive distillation. In view of the importance of batch operations in the current market-driven scenario, the present study aims to explore different options of process intensification in batch mode for a particular class of reactions.
Consider a class of reactions that
-
(a)
are reversible,
-
(b)
are characterized by the presence of azeotropes,
-
(c)
have the minimum boiling azeotrope incorporating the desired product and a reactant, and
-
(d)
the next higher boiling cut is either the pure desired product or another azeotrope consisting of a desired product of higher purity than the first azeotrope.
Esterification reactions ideally fit into this category. Esterification of acetic acid with different alcohols ranging from C1 to C5 produce different range of products like methyl acetate, ethyl acetate, iso-propyl acetate and amyl acetate, with boiling point ranges from 57 to 147 °C [13]. In addition to their industrial importance, these five quaternary systems also show complex behavior with the number of azeotropes (binary and ternary) varying from 2 to 6 with possible liquid splitting. Generally, esterification reactions are reversible in nature and the conversions are limited by equilibrium and product purities are very low. Some reactions that can be considered to belong to this class of reactions are listed in Table 1 along with their azeotropic temperatures and compositions.
High conversions can be achieved in reversible reactions by bringing about a shift in the equilibrium to the right. This can be achieved by separating the product (or products) from the reaction mixture while the reaction is in progress, thereby causing a deficit in the product concentration, and therefore shifting the equilibrium to the right. Alternatively, one of the reactants can be taken in excess, and high conversions can be achieved with respect to the limiting reactant. However, this results in the excess reactant remaining in the product mixture requiring further separation.
Process intensification in batch mode for the class of reactions considered in the present study is proposed in two ways—first by considering different units to carry out the reaction, and second by using excess of one of the reactants in each of the above units. The units considered in the present study include batch reactor (base case), single-stage separation (simple reactive batch distillation using a batch reactor–condenser arrangement) with product removal, and multi-stage separation (reactive batch distillation) with product removal and partial reflux. The objective for implementation of these options is to achieve maximum conversion with highest product purity.
Methyl acetate production case study
Methyl acetate and water are produced by the esterification of methanol with acetic acid. The non-catalytic reaction is a homogeneous liquid-phase process, where the conversion of the reactants is determined by equilibrium, according the following reaction scheme:
The reaction rate is very slow and is generally enhanced by addition of a homogeneous catalyst such as sulphuric acid. Among the four species involved in the process, methyl acetate is the most volatile component, while methanol, water and acetic acid boil at higher temperatures. The boiling points of pure components along with azeotropic compositions are reported in Table 2. As is evident from the order of boiling points in Table 2, it is not possible to achieve pure methyl acetate in batch operation.
This reaction has been one of the most well-studied systems for process intensification using reactive distillation. Eastman chemical company’s methyl acetate production features a reactive distillation column, and is one of the most successful large-scale commercial applications of process intensification [14, 15]. Other reactive distillation studies reported for this system based on heterogeneously catalyzed reaction include those carried out in continuous mode [16–20], in batch mode [21] and in semi-batch mode [22–26].
Most of the studies reported on process intensification studies for methyl acetate production have considered heterogeneous catalysts, and limited studies have been reported using homogeneous catalysts. Recently, Elgue et al. [27] considered intensification of methyl acetate production in an open plate continuous reactor (OPR) using homogeneous catalyst in terms of design and operating conditions. By intensification of design and operating conditions, the reactor allows the methyl acetate production to be carried out in a fast and continuous operation, but the purity of methyl acetate obtained at reactor outlet is limited by initial composition of the reactants. Therefore, from the production point of view, the above intensified continuous process is of low interest compared to industrial process of reactive distillation which leads to high-purity methyl acetate [15].
In the present study, it is attempted to explore different options of process intensification for this system in batch mode using the homogeneous catalyst. The objective for implementation of these options is to achieve maximum conversion with high methyl acetate purity. The equilibrium constant for this reaction is about 5.0 [28] and it is independent of the temperature. The equilibrium conversion of the reaction is 69 %, when equimolar (1:1) quantities of reactants are used (base case), and the product purity corresponds to a methyl acetate weight fraction of only 0.55.
As mentioned in the previous section, process intensification for this system aims to increase the conversion and product purity in two ways, namely by considering different units to carry out the reaction, and secondly using excess of methanol in each of the above units. The units considered are batch reactor to serve as the base case, and process intensification of integrated reaction–separation by simple reactive batch distillation (using a batch reactor–condenser arrangement) and by a multi-stage reactive batch distillation column with partial reflux. The latter option is expected to achieve better separation. However, the minimum boiling azeotrope, which has methyl acetate and methanol (0.82 and 0.18 wt fractions) as its constituents is being removed continuously, and the conversion is limited by methanol available for reaction when equimolar quantities of acetic acid and methanol are employed. Therefore, use of excess methanol is also implemented in combination with the integrated reaction–separation operation. Experimental studies are conducted in different batch units to evaluate these options of process intensification.
Materials
Methyl acetate (purity >99.436 %), methanol (purity >99.811 %), acetic acid (purity >99.746 %) and sulfuric acid (purity >98 %) are purchased from SD Fine Chemicals Ltd., and used as such.
Apparatus
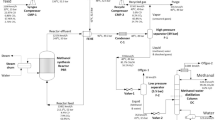
Two different batch operating units are considered to carry out the experimentation. Initially, experiments are performed in a jacketed batch reactor [28] with condenser assembly to study the base case (batch reactor) as well as simple reactive batch distillation operation (Single-stage operation). A pictorial representation of the experimental setup is shown in Fig. 1. The reactor is a 1-L glass-jacketed vessel equipped with digital overhead stirrer (direct controlled model) with a motor rating of 83/75 W, maximum torque of 85 N-cm and a speed range of 200–3,000 rpm. The impeller used is a propeller made of Teflon. The dimensions of the impeller are 5 cm diameter, blade length 1.25 cm, blade width 1 and 0.1 cm shaft diameter. The agitator is located at 3.17 cm from the bottom of the vessel (one-third of the reactor diameter). The reactor assembly is provided with a four-necked glass lid with provision for liquid-phase temperature measurement in the reactor and periodic withdrawal of liquid samples during the course of the reaction. To one of the outlets, a condenser is connected wherein cold water is circulated to minimize losses, and it is also used to collect the distillate samples during the simple reactive batch distillation operation. Temperature is measured using a Chromel Alumel type thermocouple. A Polyscience refrigerating and heating circulator thermostat model 9702 with digital temperature controller is used to maintain the temperature (±0.01 °C accuracy) in the jacketed reactor.

Schematic representation of the reactor with condenser assembly (R jacketed reactor/reboiler, TI thermo well and temperature indicator, SW sample withdrawal provision, C condenser, DW distillate samples withdrawal provision, S stirrer, TS thermostat for coolant circulation, TIC temperature indicator and controller)
The second experimental setup, as shown in Fig. 2, to explore integrated reaction–separation using multi-stage reactive batch distillation, consists a column of 1,500 mm length and 40 mm diameter with 15 bubble cap trays, a reboiler of 5-L capacity, a condenser of 1-L capacity, a reflux divider and product collector (2-L capacity). The system is equipped with temperature sensors (K-type thermocouples) in the reboiler, top tray and at different locations along the column. The contents in the reboiler are heated by a heating mantle (2 KW capacity) that has provision for heat input regulation. The column is thermally insulated. Chilled water is circulated in the condenser using a cryostat.

Schematic representation of the reactive batch distillation (RBD) column
Experimental procedure
To study the base case, the experimental setup shown in Fig. 1, is used with an overhead stirrer to mix the contents, and isothermal operation is maintained by circulating liquid using refrigerating and heating circulator. The first experiment is conducted by charging the reactor with equimolar quantities of methanol and acetic acid and subsequent addition of sulfuric acid catalyst (2 % with respect to the weight of acetic acid). The reaction is carried out at 40 °C (without condensation of the product) to serve as a base case for comparison. The reaction is stopped when there is no change in the reactor composition.
The next two experiments are conducted in a simple batch distillation assembly with an overhead condenser arrangement; the experimental setup is shown in Fig. 1. The vapor generated is condensed and collected as different cuts at different reboiler temperatures for condensate analysis. Sufficient heat input is supplied to the reboiler to maintain the reactants in boiling condition. Using two different molar ratios of acetic acid to methanol as 1:1 and 1:1.7, along with the sulfuric acid catalyst (2 % with respect to the weight of acetic acid) added to the reboiler, the reaction is carried out at the boiling point of the mixture, and the product is condensed and collected. The reaction is continued until no further collection of condensate is possible. After the reaction is completed the reboiler contents are cooled, weighed and analyzed.
The last two experiments are conducted in a multi-stage reactive batch distillation column shown in Fig. 2, using molar ratios of acetic acid to methanol as 1:1 and 1:1.7, along with the sulfuric acid catalyst (2 % with respect to the weight of acetic acid) added to the reboiler. The heat input and reflux ratio are chosen arbitrarily. The heating mantle is set at a constant heat input of 40 % of 2 KW heater capacity. After the mixture starts boiling, the vapor rises through the column and is condensed at the top of the column and part of it (25 % of the total vapor generated) is refluxed back into the column, and the remaining (75 % of the total vapor generated) is collected as a distillate product. Different cuts are collected at different top tray temperatures and samples are set aside for analysis. The reaction is continued until no further collection of condensate is possible. At the end of the run, reboiler sample is used for analysis of the unreacted acetic acid.
Analytical methods
The reaction system considered in the present study consists of esterification of methanol with acetic acid to produce methyl acetate in the presence of sulphuric acid as the catalyst. The products obtained in the reboiler and as distillate product are analyzed using different methods. The reactor/reboiler composition is determined by means of volumetric titration using NaOH with the help of predetermined calibration curves. The method of analysis is based on acid–base titrations, and therefore, it is expected that presence of sulfuric acid in the sample (being the catalyst) is likely to influence the titre value and the composition determined. Therefore, during calibration, two sets of experiments are conducted. The first set consists of preparation of synthetic reaction mixtures at different levels of conversion with respect to acetic acid. The second set consists of preparation of the same set of synthetic mixtures with the proportionate quantity of sulfuric acid based on 2 % sulfuric acid concentration with respect to initial acetic acid used. Two calibration curves are prepared based on titrating the reaction mixtures of both these sets of experiments with NaOH, and it is found that there is negligible change in both the calibration curves. The reason for this could be due to a complex formation of sulfuric acid with acetic acid delaying the ionization, thereby resulting in similar titer values in the presence and absence of sulfuric acid.
The distillate compositions of different cuts are analyzed using gas chromatography (GC-2014 Shimadzu) with TCD to analyze the composition of the distillate samples. A ZB WAX capillary column is used to separate distillate samples and to quantitatively estimate the compositions of methanol, methyl acetate and water. The GC temperature is programmed to rise from initial temperature of 50 °C with ramping @ 15 °C/min till 80 °C, and @ 25 °C/min to 130 °C, and then held constant for an additional 1.5 min. Helium gas 99.995 % is used as a carrier gas. The gas flow rate is 12 mL/min. Calibration charts of all the four components are prepared. Response factors are determined by taking synthetic mixtures of different compositions.
Results and discussion
As reported in the previous section, five experiments are conducted for the present evaluation study. The first experiment is conducted with equimolar quantities of reactants in a batch reactor at 40 °C (without condensation of the product) to serve as a base case for comparison. The equilibrium conversion obtained is 69 %. Even though the reaction is exothermic in nature and equilibrium constant is found to be almost constant with temperature, and therefore the equilibrium conversion is almost constant at different temperatures. The purity obtained corresponds to the equilibrium composition (wt fractions—methyl acetate: 0.555, methanol: 0.108, water: 0.135, acetic acid: 0.202). The next two experiments are conducted in simple reactive batch distillation assembly using molar ratios of acetic acid to methanol as 1:1 and 1:1.7, whereas the last two experiments are conducted in the multi-stage reactive batch distillation column for the same set of molar ratios. The equipment mentioned in the previous section is employed and the procedure reported earlier is followed. The results obtained in all the cases are summarized in Table 3. The compositions of different condensate cuts in the simple reactive batch distillation operation for two molar ratios along with the reboiler temperatures show that for a feed molar ratio of 1:1, 80 % conversion is achieved, whereas when the molar ratio is increased to 1:1.7, the conversion increased to 90 % with respect to acetic acid.
Table 3 also illustrates that although conversion has improved with use of excess methanol, purity with respect to methyl acetate in the latter case has decreased almost by 18 %. It can be observed that two azeotropic mixtures exist at boiling points lower than the pure products as evident from Table 2. The product compositions in Table 3 show that they do not correspond to one single azeotrope, but are a combination of both the azeotropes. Moreover, the minimum boiling azeotrope consists of methyl acetate and methanol as its components. Therefore, when methanol is available, methyl acetate combines with methanol to generate the minimum boiling azeotrope, which is of lower purity with respect to methyl acetate. Table 3 clearly shows that the first azeotrope is dominant when excess methanol is used, whereas with equimolar reactants, reasonable quantity of second azeotrope is present in the product. This can be justified by the fact that methanol available for the formation of first azeotrope is limited in case of equimolar reactant quantities.
Operation in a simple reactive batch distillation is equivalent to a single stage for separation and does not have provision for refluxing part of the condensate product to enhance separation. Therefore, the next two experiments are conducted in a multi-stage reactive batch distillation column with 15 trays and partial reflux (25 %). The results obtained during these two experiments with 1:1 and 1:1.7 molar ratios are also reported in Table 3. The conversions obtained in a multi-stage reactive batch distillation for 1:1 and 1:1.7 molar ratios are found to be 89 and 100 %, respectively. The purity of product with respect to methyl acetate is found to be 0.89 in the equimolar case, whereas it dropped to 0.70 when excess methanol was used. The reason for this decrease has already been discussed earlier. Compared to the performance in a single stage, the conversions obtained in multi-stage operation are found to be considerably increased, whereas there is only marginal improvement with respect to product purity. Higher conversions can be explained by the increased separation that can be achieved in multi-stage operation, thereby leading to a greater shift in equilibrium to the right compared to single-stage operation. The product purity obtained is comparable due to different operating times; however, the quantity of distillate collected in multi-stage operation is higher than in single-stage operation.
The results obtained in all the five experiments, as illustrated in Table 3, show comparison of all the cases using three different batch units and two feed molar ratios. As discussed earlier, the increase in conversions and decrease of methyl acetate purity with increase in methanol in the initial reaction mixture for cases 2 and 3 is evident, as we move from left to right in Table 3, whereas there is negligible change in product purity in case 1. This can be explained by the fact that no boiling of the mixture is involved in this case, and hence the presence of different azeotropes does not arise. Comparing the three cases for each molar ratio, it is clear that the conversions as well as product purities progressively increase from case 1 to case 3. This improvement is due to the incorporation of process intensification options of single-stage integrated reaction–separation in case 2 and multi-stage integrated reaction–separation with partial reflux in case 3. Therefore, this study illustrates that it is possible to achieve a trade-off between conversion and purity of methyl acetate, leading to high conversion with reasonably high product purity, in a multi-stage reactive batch distillation using optimized quantity of reactant methanol.
The analysis of results obtained above for methyl acetate case study can easily be extended for the class of reactions considered in the present study. While carrying out reactive-separation (single-stage/multi-stage), there are two factors playing opposing roles in achieving high conversion and high product purity. Based on condition (c) reported in “Process intensification schemes in batch mode”, part of the reactant also leaves along with the desired product as the first product cut, and therefore stoichiometric quantities of reactants cannot result in 100 % conversion with respect to the reactants. The product obtained is a combination of the first and second azeotropes [from conditions (c) and (d) reported in “Process intensification schemes in batch mode”], and the product purity is expected to be in between the compositions of the two azeotropes. On the other hand, if the reactant constituting the first azeotrope is taken in excess, 100 % conversion can be achieved with respect to the limiting reactant. However, the product obtained is dominated by the first azeotrope, and therefore the product purity is lower compared to the case where stoichiometric quantities of reactants are taken.
Conclusions
Different process intensification options in batch mode are proposed for a class of reactions, and are evaluated with respect to enhancement in conversion and product purity through experimental analysis for methyl acetate formation case study in different batch operating units and through use of different molar ratios of reactants. Compared to base case conversion of 69 % for 1:1 molar ratio and 83.8 % for 1:1.7 molar ratio of reactants, and a product purity of just above 0.5 wt fraction of methyl acetate, significant improvement is achieved using simple reactive batch distillation assembly (1:1 molar ratio: 80 % conversion, 0.87 wt fraction of methyl acetate; 1:1.7 molar ratio: 90 % conversion, 0.68 wt fraction methyl acetate), and in a multi-stage reactive batch distillation column (1:1 molar ratio: 89 % conversion, 0.89 wt fraction methyl acetate; 1:1.7 molar ratio: 100 % conversion, 0.70 wt fraction methyl acetate). It is evident that use of 1:1.7 molar ratios of reactants results in almost 100 % conversion at the expense of methyl acetate purity. This study illustrates that it is possible to achieve a trade-off between conversion and purity of methyl acetate, leading to high conversion with reasonably high product purity, in a multi-stage reactive batch distillation using optimized quantity of reactant methanol. The present study considers a typical case of esterification reaction with azeotropic mixtures, and the analysis is applicable to a class of reactions satisfying the criteria mentioned. Future studies will focus on finding the optimum values of the heat input and reflux ratio to achieve high conversion as well as high purity of the methyl acetate with the help of dynamic modeling, simulation and experimental studies.
Abbreviations
- k 1 :
-
Esterification reaction rate constant
- k −1 :
-
Hydrolysis reaction rate constant
- T :
-
Temperature
- TCD:
-
Thermal conductivity detector
- Q c :
-
Condenser load
- Q R :
-
Heat load to the reboiler
- D :
-
Distillate
- TI:
-
Temperature indicator
- HD:
-
Amount of distillate in the receiver
- H :
-
Amount of liquid mixture in the reboiler
- x :
-
Reboiler composition
- x D :
-
Distillate composition
- PI:
-
Process intensification
References
Charpentier JC (2007) In the frame of globalization and sustainability, process intensification, a path to the future of chemical and process engineering (molecules into money). Chem Eng J 134:84–92
Stankiewicz AI, Moulijn JA (2000) Process intensification: transforming chemical engineering. Chem Eng Prog 96:22–34
Gogate PR (2008) Cavitational reactors for process intensification of chemical processing applications: a critical review. Chem Eng Process Proc Intensif 47:515–527
Marquardt W, Kossack S, Kraemer K (2008) A frame work for the systematic design of hybrid separation process. Chin J Chem Eng 16:333–342
Maddikeri GL, Pandit AB, Gogate PR (2012) Intensification approaches for biodiesel synthesis from waste cooking oil: a review. Ind Eng Chem Res 51:14610–14628
Gole VL, Gogate PR (2012) A review on intensification of synthesis of biodiesel from sustainable feed stock using sonochemical reactors. Chem Eng Process Proc Intensif 53:1–9
Harmsen GJ (2007) Reactive distillation: The front-runner of industrial process intensification: a full review of commercial applications, research, scale-up, design and operation. Chem Eng Process 46:774–780
Sharma MM, Mahajani SM (2003) Industrial applications of reactive distillation. In: Sundmacher K, Kienle A (eds) A review in “Reactive distillation: status and future directions”. Wiley VCH, Germany, pp 1–29
Hiwale RS, Bhate NV, Mahajan YS, Mahajani SM (2004) Industrial applications of reactive distillation: recent trends. Int J Chem React Eng 2:1–53. Review no R1
Taylor R, Krishna R (2000) Modeling reactive distillation. Chem Eng Sci 55(22):5183–5229
Bollyn MP, Wright AR (1998) Development of a process model for a batch reactive distillation—a case study. Comput Chem Eng 22(1):S87–S94
Gadewar SB, Malone MF, Doherty MF (2000) Selectivity targets for batch reactive distillation. Ind Eng Chem Res 39:1565–1575
Tang YT, Chen YW, Huang HP, Yu CC (2005) Design of reactive distillation for acetic acid esterification. AIChE J 51(6):1683–1699
Agreda VH, Partin LR (1984) Reactive distillation process for the production of Methyl acetate. US Patent: 4, p 435
Agreda VH, Partin LR, Heise WH (1990) High purity methyl acetate via reactive distillation. Chem Eng Prog 86:40–46
Popken T, Gotze L, Gmehling J (2000) Reaction kinetics and chemical equilibrium of homogeneously and heterogeneously catalyzed acetic acid esterification with methanol and methyl acetate hydrolysis. Ind Eng Chem Res 39:2601
Noeres C, Kenig EY, Górak A (2003) Modeling of reactive separation processes: reactive absorption and reactive distillation. Chem Eng Process 42(3):157–178
Thery R, Meyer XM, Joulia X, Meyer M (2005) Preliminary design of reactive distillation columns. Chem Eng Res Des 83(4):379–400
Kumar MVP, Kaistha N (2009) Evaluation of ratio control schemes in a two-temperature control structure for a methyl acetate reactive distillation column. Chem Eng Res Des 87(2):216–225
Noeres C, Dadhe K, Gesthuisen R, Engell S, Górak A (2004) Model-based design, control and optimization of catalytic distillation processes. Chem Eng Process 43(3):421–434
Wajge RM, Reklaitis GV (1999) RBDOPT: a general-purpose object-oriented module for distributed campaign optimization of reactive batch distillation. Chem Eng J 75(1):57–68
Kreul LU, Górak A, Dittrich C, Barton PI (1998) Dynamic catalytic distillation: advanced simulation and experimental validation. Comput Chem Eng 22(1):S371–S378
Fernholz G, Engell S, Kreul LU, Gorak A (2000) Optimal operation of reactive batch distillation column. Comput Chem Eng 24:1569–1575
Schneider R, Noeres C, Kreul LU, Górak A (2001) Dynamic modeling and simulation of reactive batch distillation. Comput Chem Eng 25(1):169–176
Engell S, Fernholz G (2003) Control of a reactive separation process. Chem Eng Process 42(3):201–210
Volker M, Sonntag C, Engell S (2007) Control of integrated processes: a case study on reactive distillation in a medium-scale pilot plant. Control Eng Pract 15:863–881
Elgue S, Devatine A, Prat LE, Cognet P, Cabassud M, Gourdon C, Chopard F (2009) Intensification of ester production in a continuous reactor. Int J Chem React Eng 7:A24
Ganesh B, Rani KY, Sathyavathi B, Venkateshwarlu Ch (2011) Development of kinetic models for acid catalyzed methyl acetate formation reaction: effect of catalyst concentration and water inhibition. Int J Chem Kinet 43:263–277
Zeng KL, Kuo CL, Chien IL (2006) Design and control of butyl acrylate reactive distillation column system. Chem Eng Sci 61:4417–4431
Schmitt M, Hasse H, Althaus K, Schoenmakers H, Gotze L, Moritz P (2004) Synthesis of n-hexyl acetate by reactive distillation. Chem Eng Process 43(3):397–409
Acknowledgments
The authors acknowledge Department of Science and Technology (DST), New Delhi, India and CSIR XII Plan Project, INDUS MAGIC for the financial support, and the first author acknowledges CSIR, New Delhi, India for supporting financially through fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Ganesh, B., Rani, K.Y., Satyavathi, B. et al. Experimental analysis in different batch operating units for process intensification: methyl acetate production case study. Int J Ind Chem 5, 85–93 (2014). https://doi.org/10.1007/s40090-014-0023-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-014-0023-7