
Abstract
The fruits of Avicennia marina are widely used for both medicine and food in Guangxi of China. As a part of our continuous effort to search for bioactive molecules from the plant, the fruits of A. marina were investigated, which has led to one new triterpenoid saponin (1) and 29 known compounds been isolated and their structures were established by using spectroscopic methods and comparing with literature data. The new triterpenoid saponin showed cytotoxicity against GSC-3# and GSC-18# with the IC50 values were 12.21 and 5.53 μg/mL respectively, and most of the known compounds had significant antioxidant capacity with the IC50 values ranging from 0.36 to 13.07 μg/mL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Functional foods not only provide variously essential nutrition to body, but also be benefit for preventing diseases and maintaining health [1, 2]. These foods especially have multiple bioactivities, such as antioxidant, anti-inflammatory, antibacterial and anticancer activities [3, 4], so people can benefit from daily consumption of them to keep from or relieve the occurrence of chronic age-related diseases or life-style diseases [5, 6]. With the benefits, people have paid more and more attention to these foods.
Avicennia marina is one of mangrove plants and widely distribute in southeastern coast in China and its fruits are often picked and eaten by local residents [7]. Water extract of the fruits has traditionally been used for the treatments of colds, larynx and dysentery [8]. And the fruits have the effects of releasing inflammatory and dieresis as a vegetable directly [9]. Thus the fruits of A. marina reasonably meet the interests of modern people’s healthy food habits. Continuation of our study on searching for more bioactive molecules from the plant had led to the isolation of one new triterpenoid saponin (1) and 29 known compounds from the fruits of A. marina. Pharmacological experiments showed that the new triterpenoid saponin had antitumor activity and most of the known compounds had potential antioxidant activity.
2 Results and Discussion
2.1 Structures Identification of Isolated Compounds
With various chromatographic methods, 30 compounds were isolated from the EtOAc fraction and n-BuOH fraction of the fruits of A. marina ethanol extract. With the basis spectroscopic data and comparison with literature data, these compounds were identified as ilekudinoside B (2) [10], ilekudinoside C (3) [11], caffeic acid (4) [12], ferulic acid (5) [13], 4-hydroxycinnamic acid (6) [14], p-hydroxybenzoic acid (7) [15], 4-ethylcatechol (8) [16], 3,4,5-trimethoxybenzoic acid (9) [17], avicennone E (10) [18], avicennone D (11) [18], glechomol C (12) [19], trans-1,3-bis(3′,4′-dihy-droxyphenyl)-1-butene (13) [20], (E)-3-(3′-hydroxybut-1-enyl)-2,4,4-trimethylcyclohexa-2,5-dienone (14) [21], 3-(3′-oxobutyl)-2,4,4-trimethylcyclohexa-2,5-dienone (15) [22], 3-(3′-hydroxybutyl)-2,4,4-trimethylcyclohexa-2,5-dienone (16) [22], acteoside (17) [23], calceolarioside A (18) [24], campneoside I (19) [25], [β-D-glucopyranose] [3-O-(6-deoxy-α-l-mannopyranosyl)] [4-(2E)-3-(3,4-dihydrox-yphenyl)-2-propenoate] (20) [23], 6″-O-acetylacteoside(21) [26], crenatoside (22) [27], decaffeoylacteoside (23) [26], flavoyadorinin B (24) [28], marinoid D(25) [29], 10-O-(trans-feruloyl) geniposidic acid (26) [30], chlorogenic acid (27) [31], neochlorogenic acid(28) [31], cleroindicin E (29) [32], quinic acid ethyl ester (30) [33], and a new compound, 6′-O-(n-butanol) ilekudinoside B ester (1).
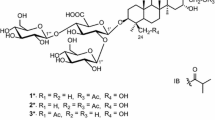
Compound 1 was obtained as white amorphous powder. Its molecular formula was determined to be C46H74O15 on the basis of its HRESIMS (m/z 889.4921 [M + Na]+, calcd C46H74O15Na, 889.4920), suggesting 10 degrees of unsaturation. The IR absorption bands at 3430 and 1735 cm−1 implied the presence of the hydroxyls and carboxyl groups, respectively. The 1H NMR spectrum of 1 exhibited signals for seven methyl groups at δH 1.27 (s), 1.07 (s), 0.97 (s), 0.85 (s), 0.75 (s), 0.66 (s) and 0.85 (d, J = 6.0 Hz) and an olefinic proton at δH 5.16 (s), which was in conformity with the appearance of seven methyls (δC 27.6, 16.3, 15.2, 16.4, 23.8, 26.4 and 16.3) and one olefinic signal (δC 127.1) in its 13C NMR data (Table 1). In addition, the presence of two anomeric carbon at δC 105.1 and 94.1, as well as other oxygenated carbon signals from δC 60.7 to 77.6 and a carboxyl at δC 169.1 in the 13C NMR spectrum, indicated that there existed sugar moiety. These data, in association with the literature investigation, suggested that compound 1 might have a structure related to that of ilekudinoside B (Fig. 1), except for the appearance of three methylenes (δC 64.1/δH 4.09, δC 30.1/δH 1.55, δC 18.5/δH 1.33) and one methyl (δC 13.6/δH 0.87), which were identified to n-butanol, and supported by the 1H–1H COSY correlations of 4.09/1.55/1.33/0.87 (Fig. 2) [34]. Comparison of the 13C NMR data of 1 with those of ilekudinoside B revealing the upfield shift (− 3.1 ppm) for C-6′, assumed that the additional n-butanol was attached to C-6′. The assumption was further supported by the HMBC correlation from H-1′″ (δH 4.09) to C-6′ (δC 169.1) (Fig. 2). The sugar analysis by GC after acid hydrolysis afforded d-glucose and d-glucuronic acid as component monosaccharides. And the coupling constants of the anomeric protons [δH 4.29 (d, J = 7.8 Hz, H-1′), 5.15 (d, J = 7.8 Hz, H-1″)] suggested β-pyranosyl configuration for both d-glucose and d-glucuronic acid moieties [35]. Moreover, in the ROESY spectrum, the ROESY correlation of δH 3.04 (H-3) with 0.72 (H-5) (Fig. 3) suggested that the configuration of H-3 was the β orientation, due to α oriented H-5 from the biosynthesis of ursane-type triterpene [36]. Meantime the ROESY correlation of δH 3.83 (19-OH) with 0.85 (30-H) suggested the configuration of 19-OH was α orientation (Fig. 3). Therefore, 1 was elucidated as 6′-O-(n-butanol) ilekudinoside B ester.

Structures of 1 and 2

Key 1H-1H COSY (blue line) and HMBC (red arrow) correlations of compound 1

Key ROESY (red arrow) correlations of compound 1
2.2 Anticancer Activity
All compounds were evaluated for their bioactivities against two human glioma stem cell lines (GSC-3# and GSC-18#) by the cell viability assay and phenotypic screening. The results showed that compound 1 exhibited the moderate cytotoxicity against GSC-3# and GSC-18# at the concentration of 20 μg/mL (Fig. 4), and the IC50 values were 12.21 and 5.53 μg/mL, respectively (Fig. 5).

Compound 1 against human glioma stem cells by phenotypic screening at 20 μg/mL

The IC50 value for compound 1 against human glioma stem cell lines
2.3 Antioxidant Activity
DPPH is a widely-used stable free radical to evaluate antioxidant activity of bioactive. In this present assay, the free radical scavenging activities of the polyphenols and phenylethanoid glycosides were evaluated and compounds 4, 5, 8, 18–23, 27, 28 showed potent scavenging activities with IC50 values from 0.36 to 13.07 μg/mL. Especially compounds 4, 8, 18, and 21 exhibited well scavenging capacities than vitamin C in the experiment (Table 2).
3 Experimental Section
3.1 General Experimental Procedures
1D and 2D NMR spectra were recorded on Bruker DRX-500 spectrometers using TMS as an internal standard. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. Optical rotations were measured on a JASCO P-1020 polarimeter. IR spectra were determined on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr disks. UV spectra were detected on a SHMADZU UV-2401PC spectrometer. MS and HRMS analysis were carried out on Waters Xevo TQS and Waters AutoSpec Premier P776 mass spectrometers, respectively. Semipreparative HPLC was performed on a Waters 600 with a COSMOSIL C18 (10 × 250 mm) column. Silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemical Co., Ltd., People’s Republic of China), and MCI gel (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Fractions were monitored by thin-layer chromatography (TLC) (GF254, Qingdao Marine Chemical Co., Ltd.), and spots were visualized by 10% sulfuric acid ethanol solution.
3.2 Plant Materials
The fresh fruits of A. marina were collected in Beibu Gulf of Guangxi province, China, and identified by Dr. Ya-Ping Liu, Kunming Institute of Botany, CAS. A voucher specimen (No. 20170402) has been deposited in the Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China.
3.3 Extraction and Isolation
The fresh fruits of A. marina (14.2 kg) were extracted with 95% EtOH (40 L) three times under ambient temperature. After removing the solvent in vacuum, the residue (300 g) was suspended in H2O and extracted successively with petroleum ether (PE), ethyl acetate (EtOAc) and n-butanol (n-BuOH) respectively. After evaporation of the solvent in reduced pressure, the PE fraction (12 g), EtOAc fraction (18 g) and n-BuOH fraction (89 g) were obtained. The EtOAc fraction were separated by C18 silica gel column eluting with CH3OH/H2O (from 40:60 to 100:0, v/v) to yield four fractions (Fr-1 to Fr-4). Fr-4 was purified by C18 silica gel column eluting with CH3OH/H2O (10:90, v/v) to afford compound 7 (4 mg) and the residue, then the latter was further purified over HPLC using the mobile phase of 30% CH3CN/H2O and compounds 10 (2.0 mg) and 11 (2.4 mg) were obtained. Fr-3 was separated by silica gel column eluting with CHCl3/CH3OH (8:1, v/v) to provide compounds 4 (10 mg) and 5 (12 mg) and fractions Fr-3-1, Fr-3-2 and Fr-3-3. Fraction Fr-3-1 then was separated by silica gel column eluting with PE/Acetone (3:1, v/v) to afford compound 9 (60 mg). Fraction Fr-3-2 were further purified by HPLC using the mobile phase of 33% CH3CN/H2O to give compounds 8 (18 mg) and 6 (16.9 mg). Compounds 14 (12 mg) and 16 (3.8 mg) from fraction Fr-3-3 by silica gel column and eluted by PE/acetone (5:1, v/v). Fr-2 was divided into two fractions (Fr-2-1 and Fr-2-2) by Sephadex LH-20 column, eluting with 100% CH3OH. And then Fr-2-1 afforded compound 15 (2.8 mg) by silica gel column eluting with PE/acetone (2:1, v/v). Fr-2-2 was purified by silica gel column eluting with CH3Cl/acetone (2:1, v/v) to afford compound 12 (10 mg). Compound 13 (2.5 mg) were obtained from Fr-1 by HPLC eluting with 20% CH3CN/H2O.
The n-BuOH fraction was separated by C18 silica gel column which eluted by CH3OH/H2O (from 10:90 to 100:0, v/v) to yield eight fractions (Fr-I to Fr-VIII). Fr-VIII was divided into three subfractions (Fr-VIII-1 to Fr-VIII-3) by silica gel column gradually eluting with CH3Cl/CH3OH (10:1, 5:1, 0:1, v/v). And then fraction Fr-VIII-3 was separated by Sephadex LH-20 CC eluting with methanol to afford compound 1 (290 mg) and the residue. Compound 3 (24.3 mg) was obtained by silica gel column eluting with CH3Cl/CH3OH (5:1, v/v) from the residue. Compound 2 (3.8 g) were purified from Fr-VIII-2 by silica gel column eluting with CH3Cl/CH3OH (4:1, v/v). Fr-VIII-1 was purified by silica gel column eluting with CH3Cl/CH3OH (3:1, v/v) to afford 18 (15.5 mg). Compounds 19 (33.0 mg) and 20 (161 mg) were obtained by silica gel column eluting with CH3Cl/CH3OH (5:1, v/v) and then HPLC with 40% CH3CN/H2O as the mobile phase from Fr-VII. Fr-VI was divided into subfractions Fr-VI-1 and Fr-VI-2. And then compounds 21 (24.2 mg) and 23 (26 mg) were obtained from Fr-VI-1 by HPLC eluting with 45% CH3CN/H2O, and Fr-VI-2 eluting with 45% CH3CN/H2O to afford compounds 22 (63 mg) and 24 (2.2 mg). Fr-V was separated by silica gel column eluting with CH3Cl/CH3OH (3:1, v/v) and then purified by HPLC eluting with 33% CH3CN/H2O to afford compounds 25 (20 mg) and 26 (7.2 mg). Fr-IV was purified by silica gel column using the mobile phase of CH3Cl/CH3OH (3:1, v/v) to afford compound 17 (162 mg). Fr-III was further divided into two subfractions (Fr-III-1 and Fr-III-2). Compounds 27 (15 mg) and 28 (35.9 mg) were obtained from Fr-III-1 by HPLC eluting with 23% CH3CN/H2O. Fr-III-2 was purified by silica gel column eluting with CH3Cl/CH3OH (8:1, v/v) to afford compounds 29 (28.4 mg) and 30 (27.7 mg).
6′-n-butanol, ilekudinoside B ester (1) White amorphous powder; \([\alpha ]_{\text{D}}^{20}\) − 20.1 (c 1.30, MeOH)]; IR (KBr) νmax 3430, 2932, 1735, 1070 cm−1; UV (MeOH) λmax (log ε) 206 (4.5) nm; HRESIMS m/z 889.4921 ([M + Na]+) (calcd for C46H74O15Na, 889.4920); 1H and 13C NMR spectroscopic data, see Table 1.
3.4 Acid Hydrolysis of Compound 1
As previously described [37] with some minor modifications, compound 1 (10 mg) was hydrolyzed with 2 M HCl/dioxane (1:1, 4 mL) under reflux for 8 h. After partitioned with CHCl3 (2 mL × 3), the aqueous layer was neutralized with 2 M NaOH and then dried to give a monosaccharide mixture. A solution of the sugar mixture in pyridine (2 mL) was added to l-cysteine methyl ester hydrochloride (about 1.5 mg) and kept at 60 °C for 1 h. Then trimethylsilylimidazole (1.5 mL) was added to the reaction mixture. After kept at 60 °C for 30 min, the reaction mixture was immediately subjected to GC analysis, run on a Shimadzu GC-14C gas chromatography equipped with an H2 flame ionization detector. The column was a 30 m × 0.32 mm i.d. 30QC2/AC-5 quartz capillary column with the following conditions: column temperature: 180–280 °C; programmed increase: 3 °C/min; carrier gas: N2 (1.0 mL/min); injector and detector temperature: 250 °C; injection volume: 4 μL; and split ratio: 1/50. By comparing with the retention time of the authentic sugars in the form of derivatives under the same condition, the sugar moieties of compound 1 was determined to be d-glucose (tR: 22.895 min), D-glucuronic acid (tR: 23.958 min) by compare with standard d-glucose (tR: 22.806 min) and D-glucuronic acid (tR: 23.907 min).
3.5 Anticancer Activity
GSC-3# and GSC-18# were human glioma stem cell lines that were established by Kunming institute of zoology from two human glioblastoma multiform samples. The glioma stem cell was cultured in serum-free medium DMEM F12 supplied with 1xB27 and 50 ng/mL EGF, BFGF and 1% penicillin/streptomycin. The glioma stem cells were seeded in the laminin pre-coating dishes and cultured in 37 °C, 5% CO2 incubator. Cell viability assay was performed by the MTS method as previously described. GSCs were digested and counted, seeded in laminin pre-coating 96-well-plate with 20000 cells/well. The compounds were added with a serial gradient concentration (20, 10, 5, 2.5, 1.25, 0.625 μg/mL) and cultured in cell incubator for 72 h. MTS reagent was diluted 1:5 with fresh medium and mixed well. The old medium was removed and subsequently the fresh medium was added with 100 μL/well. The cells were incubated for 1.5 h. Absorbance was measured by Hybrid Reader (BioTek synergy H1) at 490 nm. The cell viability was evaluated by percentage compared with DMSO control group. The half-maximal inhibitory concentration (IC50) was measured and calculated by Graph Pad Prism 5 software.
3.6 DPPH Free Radical Scavenging Activity
The antioxidant activity of compounds was evaluated using the DPPH free radical scavenging assay. The procedure used is an adaptation of those previously [38]. Samples were dissolved to various concentrations (25, 15, 5, 2, 0.5 μg/mL) with methanol. In this assay, reaction mixtures containing a methanolic solution of 0.1 mmol/L DPPH (160 μL) and serial dilutions of test samples (40 μL) were placed in a 96-well-plate. MeOH was used as a negative control and vitamin C was used as a positive control. The reactive mixtures were incubated at 37 °C for 30 min in darkness. The absorbance of the mixtures at 517 nm was measured by microplate reader. The scavenging activity (%) was determined by the following equation:
Percent inhibition was plotted against concentration, and the equation for the line was used to obtain the IC50 value.
References
S.Z. Wang, Z.L. Li, G.L. Yang, C.T. Ho, S.M. Li, Food Funct. 8, 1749–1762 (2017)
S. Mihara, H. Tateba, O. Nishimura, Y. Machii, K. Kishino, J. Agric. Food Chem. 35, 532–537 (1987)
P.F. Zhu, Z. Dai, B. Wang, X. Wei, H.F. Yu, Z.R. Yan, X.D. Zhao, Y.Y. Liu, X.D. Luo, Nat. Prod. Bioprospect. 7, 421–431 (2017)
X.R. Wang, C.L. Zhang, Y.J. Peng, H.M. Zhang, Z.G. Wang, Y.L.Y. Gao, H.L. Zhang, Food Chem. 246, 41–47 (2018)
J.M. Kong, L.S. Chia, N.K. Goh, T.F. Chia, R. Brouillard, Phytochemistry 64, 923–933 (2003)
R. Krikorian, M.D. Shidler, T.A. Nash, W. Kalt, M.R. Vinqvist-Tymchuk, B. Shukitt-Hale, J.A. Joseph, J. Agric. Food Chem. 58, 3996–4000 (2010)
X.Q. Ning, Y.B. Lin, Y.F. Tan, Guide China Med 11, 73–75 (2013)
C.L. Shao, X.M. Fu, C.Y. Wang, L. Han, X. Liu, Y.C. Fang, G.Q. Li, G.X. Liu, H.S. Guan, Period. Ocean Univ. China 39, 712–718 (2009)
W.P. Xie, C.H. Gao, X.X. Yi, W. Yi, B.J. He, B. Chen, Guihaia 34, 398–401 (2014)
K. Nishimura, T. Miyase, H. Noguchi, J. Nat. Prod. 62, 1128–1133 (1999)
L. Wang, Y. Cai, X.Q. Zhang, Carbohydr. Res. 349, 39–43 (2012)
T.J. Hsich, Y.C. Wu, W.L. Lo, S.C. Chen, S.H. Kuo, C.Y. Chen, Chin. Pharm. J. 56, 17–23 (2004)
C.N. Xia, H.B. Li, F. Liu, W.X. Hu, Bioorg. Med. Chem. Lett. 18, 6553–6557 (2008)
Y.M. Chang, H.K. Liu, J.M. Lo, S.C. Chien, Y.F. Chan, T.H. Lee, J.K. Su, Y.H. Kuo, J. Chin. Chem. Soc. 50, 161–166 (2003)
H. Saito, M. Yokoi, M. Aida, M. Kodama, T. Oda, Y. Sato, Magn. Reson. Chem. 26, 155–161 (1988)
R. Lang, C. Mueller, T. Hofmann, J. Agric. Food Chem. 54, 5755–5762 (2006)
A. Alam, S. Tsuboi, Tetrahedron 63, 10454–10465 (2007)
L. Han, X.S. Huang, H.M. Dahse, U. Moellmann, H.Z. Fu, S.S. Grabley, I. Sattler, W.H. Lin, J. Nat. Prod. 70, 923–927 (2007)
Q.F. Zhu, Y.Y. Wang, W. Jiang, H.B. Qu, J. Asian Nat. Prod. Res. 15, 258–264 (2013)
O. Frank, S. Blumberg, C. Kunert, J. Agric. Food Chem. 55, 1945–1954 (2007)
V. Milovanovic, N. Radulovic, V. Mitic, R. Palic, G. Stojanovic, J. Essent. Oil Res. 20, 310–314 (2008)
A.F. Barrero, E.J. Alvarez-Manzaneda, R. Chahboun, A.R. Rivas, P.L. Palomino, Tetrahedron 56, 6099–6113 (2000)
T. Tanaka, T. Ikeda, M. Kaku, X.H. Zhu, M. Okawa, K. Yokomizo, M. Uyeda, T. Nohara, Chem. Pharm. Bull. 52, 1242–1245 (2004)
Y.J. Chen, H.G. Zhang, X. Li, Chem. Nat. Compd. 45, 330–332 (2009)
M.Z. Yang, X.Q. Wang, C. Li, Chin. Tradit. Herb. Drugs 17, 2447–2452 (2014)
T. Kanchanapoom, R. Kasai, K. Yamasaki, Phytochemistry 59, 565–570 (2002)
T. Shen, X.Q. Li, W.C. Hu, J. Korean Soc. Appl. Biol. Chem. 58, 617–625 (2015)
E.J. Lee, J.S. Kim, H.P. Kim, J.H. Lee, S.S. Kang, Food Chem. 120, 134–139 (2010)
Y. Sun, J. Ouyang, Z.W. Deng, Q.S. Li, W.H. Lin, Magn. Reson. Chem. 46, 638–642 (2008)
Y.J. Narukawa, N. Shimizu, T. Takeda, Nat. Med. 55, 79–82 (2001)
T. Han, H.L. Li, Q.Y. Zhang, H.C. Zheng, L.P. Qin, Chem. Nat. Compd. 42, 567–570 (2006)
J. Tian, Q.S. Zhao, H.J. Zhang, J. Nat. Prod. 60, 766–769 (1997)
X.J. Shi, J.H. Zhu, L. Yang, Chin. Med. Mat. 42, 870–873 (2011)
I.C. Jones, G.J. Sharman, J. Pidgeon, Magn. Reson. Chem. 43, 497–509 (2005)
B. Wang, Z. Dai, L. Liu, X. Wei, P. Zhu, H.F. Yu, Y.P. Liu, X.D. Luo, Nat. Prod. Bioprospect. 6, 285–290 (2016)
K.O. Eyong, G. Bairy, A.A. Eno, J. Taube, K.G. Hull, G.N. Folefoc, H.S. Foyet, D. Romo, Med. Chem. Res. 27, 268–277 (2018)
Y.F. Qu, J.Y. Gao, J. Wang, Y.M. Geng, Y. Zhou, C.X. Sun, F. Li, L. Feng, M.J. Yu, G.S. Wang, Molecules 22, 1731/1–1731/9 (2017)
S.M. Kazuko, F.K. Kuniko, Y.H. Keiko, N.K. Takashi, J. Agric. Food Chem. 40, 945–948 (1992)
Acknowledgements
The authors are grateful to the Natural Science Foundation of China (31770388, 81225024) and the “Ten Thousand Plan”, a National High-level Talents Special Support Plan for partial financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
13659_2018_167_MOESM1_ESM.docx
1D and 2D NMR spectra, HRESIMS, IR and UV spectra and Optical rotation of compound 1 are available as Supplementary Information. Supplementary material 1 (DOCX 978 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, XW., Dai, Z., Wang, B. et al. Antitumor Triterpenoid Saponin from the Fruits of Avicennia marina. Nat. Prod. Bioprospect. 8, 347–353 (2018). https://doi.org/10.1007/s13659-018-0167-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-018-0167-9