
Abstract
Phytochemical reinvestigation on the cultural broth of Boreostereum vibrans led to the isolation of six new vibralactone derivatives, vibralactone N (1), vibralactone O (2), vibralactone P (3), 10-lactyl vibralactone G (4), (3S*, 4R*)-6-acetoxymethyl-2,2-dimethyl-3,4-dihydro-2H-chromene-3,4-diol (5), vibralactone Q (6). Their structures were elucidated by extensive spectroscopic methods.
Graphical Abstract

.
Similar content being viewed by others

1 Introduction
Boreostereum vibrans (synonym Stereum vibrans) is a fungus belonged to the family Boreostereaceae which is characterized by possessing diverse bioactive compounds [1–7]. Vibralactone, first reported in 2006, is a rare fused β-lactone isolated from B. vibrans with significant lipase inhibitory activity (IC50 = 0.4 μg/mL) [8]. This distinguished compound has aroused many follow-up studies. In 2008, first total synthesis of vibralactone was reported [9, 10]. In 2011, vibralactone was used as a tool to study the activity and structure of the ClpP1P2 complex from Listeria monocytogenes was published [11]. Our continuous investigations on the chemical constituents of the culture of B. vibrans have led to a series of reports on bioactive vibralactone derivatives [12–15]. Recently the biosynthetic origin of vibralactone and its biosynthetic pathway which includes several very interesting reactions were established [16]. In order to explore and understand the potential for the production of secondary metabolites by B. vibrans, a scale-up fermentation of this fungus was carried out. Very careful investigation of the culture has resulted in the isolation of six new vibralactone derivatives. This paper deals with the isolation and structure elucidation of these compounds.
2 Results and Discussion
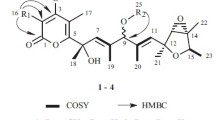
Compound 1 was isolated as a colorless oil and determined to have a molecular formula of C13H20O4 based on the HREIMS data, m/z 240.1361 [M]+ (calcd for 240.1362). The IR absorption bands at 3430, 1729 and 1634 cm−1 suggested the presence of hydroxyl, carboxyl and double bond groups, respectively. From its 1H NMR spectrum, two olefinic protons (δH 5.06 and 5.61) can obviously be found, as well as three methyl groups at δ 1.57 (3H, s), 1.65 (3H, s) and 3.61 (3H, s). The 13C NMR and DEPT spectra of 1 showed thirteen carbons resonances, including three methyls, three methylenes, four olefinic carbons, an oxymethine, and two quaternary carbons (δC 65.8, C-1; 174.5, C-7; Table 1). Detailed analyses of the 2D NMR spectra of 1 revealed that it was similar to those of vibralactone E [12]. In the HMBC spectrum of 1, an obvious correlation was observed from a methoxy (δH 3.61) to the carboxyl group (δC 174.5, C-7). To draw a conclusion, compound 1 was esterified by a methyl at the carboxyl group of vibralactone E. The relative configuration of 1 was determined by a ROESY experiment. In the ROESY spectrum, H-5 correlated to the two protons of C-8, indicating that both H-5 and the isopentenyl group were β oriented. Compound 1 was named vibralactone N (Fig. 1).

Structures of compounds 1–6
Compound 2, a colorless oil, was determined to have a molecular formula of C12H20O3 according to the HREIMS data, m/z 212.1404 [M]+ (calcd for 212.1412). The IR spectrum revealed the presence of hydroxyl (3388 cm−1) and carboxyl (1735 cm−1) groups. The 1D NMR spectra demonstrated twelve carbons, which were ascribed to two methyls, four methylenes, four methines and two quaternary carbons (Table 1). These spectroscopic data showed that 2 was very similar to those of vibralactone I [14]. The analyses of 1H-1H COSY and HMBC spectra of 2 suggested that 2 possessed a same planar structure with that of vibralactone I. Its 13C NMR and DEPT spectra showed that the chemical shifts of C-4 (δC 31.0) and C-5 (δC 50.8) of 2 were downfield shifted, while C-1 (δC 218.7) was upfield shifted obviously comparing to the corresponding signals of vibralactone I (C-1, δC 220.2, C-4, δC 28.3; C-5, δC 47.3). These data suggested that 2 was a stereoisomer of vibralactone I. In the ROESY spectrum, a key correlation of H-4 with H-8 and strong correlations from H-8 to H-6 and H-7 suggested H-2, H-3 and H-5 were in the same side (Fig. 2). Therefore, the structure of 2 was elucidated and named vibralactone O (Fig. 1).

Selected 2D NMR correlations of compounds 2 and 4
The molecular formula of compound 3 was established as C12H18O3 by HREIMS (m/z 210.1250, calcd for 210.1256). Compared its 13C NMR and DEPT spectroscopic data with those of compound 2, the chemical shifts of the carbons in 3 were similar to the corresponding carbons in 2, with the exception of chemical shifts of C-2 (δC 137.8) and C-3 (δC 173.2). Subsequent analysis of the 2D NMR spectroscopic data of 3 suggested the existence of an α,β-unsaturated ketone, owing to C-2 and C-3 were oxidized to form a double bond which conjugated with the carbonyl group (δC 210.7). The stereo-configuration of C-5 was not determined currently. Compound 3 was named vibralactone P (Fig. 1).
Compound 4 was obtained as a colorless oil. Its molecular formula was determined as C13H20O5 by HRESIMS (m/z 279.1203 [M + Na]+, calcd for 279.1208), with four degrees of unsaturation. The IR spectrum revealed the existence of hydroxy (3437 cm−1) and carboxyl (1766 cm−1) groups. The 13C NMR and DEPT spectra showed thirteen carbons, including three methyls, three methylenes, four methines (three were oxygenated) and three quaternary carbons (two olefinic carbons and two lactone carbons; Table 2). In the HMBC spectrum, the proton at 1.34 ppm (d, J = 6.6 Hz, H-3′) was correlated to a methine (δC 67.5, C-2′) and a carbonyl group (δC 175.4, C-1′), as well as cross peaks from 2′-OH (δH 4.24) and H-3′ to H-2′ (δH 4.25) in the COSY spectrum, revealed that the presence of a lactic acid group. Further analyses of the 2D NMR spectroscopic data of 4 suggested that the other parts of 4 were similar to those of vibralactone G both in planar structure and stereo-configuration [14]. From the HMBC spectrum, significant correlations were observed from H-10 (δH 4.51, dd, J = 12.4, 2.8 Hz; 4.55, dd, J = 12.4, 2.8 Hz) to the lactic carbonyl group (δC 175.4, C-1′). These signals confirmed that 4 was a lactyl-substituted derivative of vibralactone G. The relative configuration was determined by a ROESY experiment. The correlations from H-3 (δH 2.79, m) to H-6 (δH 1.31, d, J = 6.4 Hz) were found in the ROESY spectrum. We have not observed the cross peaks from H-3 to H-5 (δH 4.65, m) in the ROESY. It suggested that H-3 and Me-6 (δC 21.3) were in the same side. On the other hand, strong cross peaks were observed from H-8 (5.53, br. t, J = 7.8 Hz) to H-10 (4.51, dd, J = 12.4, 2.8 Hz; 4.55, dd, J = 12.4, 2.8 Hz). These evidences suggested that the double bond (C8–C9) was a E configuration (Fig. 2). Therefore, compound 4 was identified as 10-lactyl vibralactone G, as shown in Fig. 1.
Compound 5 was isolated as a pale yellow solid. It had a molecular formula as C14H18O5 from the HREIMS (m/z 266.1147 [M]+, calcd for 266.1154). In the 1H NMR spectrum, the ABX spin system observed from aromatic protons at δH 7.48 (1H, d, J = 2.1 Hz), 7.16 (1H, dd, J = 8.3, 2.1 Hz), and 6.70 (1H, d, J = 8.3 Hz) reveals the presence of a 1,2,4-trisubstituted benzene ring. Besides, in the 1H-1H COSY spectrum, cross peaks from two hydroxyl protons at δH 4.60 (1H, d, J = 6.1 Hz) and 4.67 (1H, d, J = 4.6 Hz) to H-3 (δH 3.53, dd, J = 8.4, 6.1 Hz) and H-4 (δH 4.51, dd, J = 8.4, 4.6 Hz), respectively, as well as cross peaks from H-3 to H-4 were observed, these data suggested the existence of a 1,2-diols group. The coupling constant of H-3/4 (J = 8.4 Hz) suggested that the two hydroxyl groups were in the opposite side. The HMBC spectrum showed that H-11 (δH 4.99, s, 2H) exhibited a clear correlation to the carboxyl group (δC 170.9, C-1′). Meanwhile, only one signal could be detected from the methyl singlet at δH 2.01 ppm to the carbonyl group. All these evidences suggested that 5 was an acetylation derivative of (3S*,4R*)-6-(hydroxymethyl)-2,2-dimethyl-3,4-dihydro-2H-chromene-3,4-diol at the position of hydromethyl group. (3S*,4R*)-6-(Hydroxymethyl)-2,2-dimethyl-3,4-dihydro-2H-chromene-3,4-diol was isolated from the fermentation broth of a marine sediment-derived fungus Eutypella scoparia FS26 obtained from the South China Sea as one of the two new polyketides [17]. Thus, compound 5 was established as (3S*,4R*)-6-acetoxymethyl-2,2-dimethyl-3,4-dihydro-2H-chromene-3,4-diol, as shown in Fig. 1.
Compound 6, a colorless oil, was determined to have a molecular formula of C12H18O3 based on the HREIMS data, m/z 210.1259 [M]+ (calcd for 210.1256). The strong adsorption bands at 3427 and 1717 cm−1 suggested the presence of hydroxyl and carboxyl groups. The 1D NMR spectroscopic data demonstrated twelve carbons signals, including four olefinic carbons and a carbonyl. According to the HMBC spectrum, correlations can be found from both two methyl singlets (δH1.62, H-10; 1.70, H-11) to two olefinic carbons (C-8, C-9). Meanwhile, cross peaks from H-7 to H-8 were also displayed in the 1H-1H COSY spectrum. These data confirmed the presence of an isopentenyl unit. Furthermore, the 1H-1H COSY correlations established connections from C-2/C-3/C-12/C-13. The HMBC spectrum showed that H-7 (δH 2.92) correlated to C-1, C-2 and C-6, H-2 (δH 6.67, d, J = 3.9 Hz) correlated to C-4 and C-6, as well as H-4 (δH 4.11, 1H, dd, J = 10.8, 7.2 Hz; 4.37, 1H, dd, J = 10.8, 4.8 Hz) correlated to C-6. The stereo-configuration of C-3 was not determined currently. Compound 6 was named vibralactone Q (Fig. 1).
3 Experimental Section
3.1 General Experimental Procedures
UV spectra were obtained using a Shamashim UV 2401 spectrometer. Optical rotations were recorded on a JASCO P-1020 polarimeter. IR spectra were measured on a Bruker Tensor-27 infrared spectrophotometer with KBr pellets. HREIMS were obtained on a Waters Autospec Premier P776 mass spectrometer. HRESIMS were taken on an Agilent G6230 TOF MS spectrometer. 1D and 2D NMR spectra were recorded on Bruker Avance-600 and Ultrashield-800 spectrometers using TMS as an internal standard. Silica gel 200–300 mesh (Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for column chromatography. Medium pressure liquid chromatography (MPLC) was performed on a Büchi Sepacore System equipping pump manager C-615, pump modules C-605 and fraction collector C-660 (Büchi Labortechnik AG, Switzerland), and columns packed with Chromatorex C-18 (40–75 μm, Fuji Silysia Chemical Ltd., Japan). Preparative HPLC was performed on an Agilent 1260 liquid chromatography system equipped with a Zorbax SB-C18 column (5 μm, 9.4 mm × 150 mm).
3.2 Fungus Material and Cultivation Conditions
The fungus B. vibrans was provided and fermented by Zheng-Hui Li, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. 20120920B) was deposited at the Herbarium of Kunming Institute of Botany. The culture medium to ferment this fungus consist of glucose (5 %), peptone from porcine meat (0.15 %), yeast powder (0.5 %), KH2PO4 (0.05 %) and MgSO4 (0.05 %). Five hundred 500-mL Erlenmeyer flasks each containing 350 mL of above-mentioned culture medium were inoculated with B. vibrans strains, respectively. Then they were incubated on rotary shakers at 24 °C and 150 rpm for 25 days in dark environment.
3.3 Extraction and Isolation
The culture broth (400 L) of B. vibrans was filtered, and the filtrate was extracted four times with ethyl acetate (EtOAc). Meanwhile, the mycelium was extracted by CHCl3/MeOH (1:1) for three times. The EtOAc layer together with the mycelium extraction was concentrated under reduced pressure to afford a crude extract (353 g). Then this residue was subjected to column chromatography over silica gel (200–300 mesh) eluting with a gradient of petroleum ether/acetone (100:0 → 0:100) to give five fractions (A–E). Fraction B was separated by MPLC eluting with (MeOH/H2O, 10:90 → 100:0) to afford ten subfractions (B1–B10). Subfraction B4 was subjected to normal phase column chromatography eluting with petroleum ether/acetone (4:1 → 2:1) to give 3 (0.5 mg). Subfraction B5 was subjected to reverse phase column chromatography eluting with MeOH/H2O (40:60 → 60:40) followed by Sephadex LH-20 (acetone) column chromatography to yield 1 (3.5 mg) and 2 (1.8 mg). Subfraction B7 was separated by preparative HPLC (MeCN/H2O, 15/85, 10 mL/min), and then purified by Sephadex LH-20 (acetone) column chromatography to afford 4 (2.3 mg) and 5 (0.8 mg). Subfraction B8 was purified by preparative HPLC (MeCN/H2O, 20/80, 10 mL/min) to yield 6 (3.8 mg).
3.4 Vibralactone N (1)
Colorless oil; [α]+5.3 (c 0.12, MeOH). UV (MeOH) λmax nm (log ε): 202 (3.03). IR (KBr) νmax cm−1: 3430, 2964, 2924, 2857, 1729, 1634, 1439, 1384, 1233, 1057. For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Table 1. HREIMS m/z: 240.1361 [M]+ (calcd for C13H20O4, 240.1362).
3.5 Vibralactone O (2)
Colorless oil; [α]+82.8 (c 0.05, MeOH). UV (MeOH) λmax nm (log ε): 203 (3.60), 302 (1.94). IR (KBr) νmax cm−1: 3388, 2964, 2924, 2878, 1735, 1452, 1378, 1154, 1075, 1053, 1027. For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Table 1. HREIMS m/z: 212.1404 [M]+ (calcd for C12H20O3, 212.1412).
3.6 Vibralactone P (3)
Yellow oil; [α]−24.0 (c 0.02, MeOH). UV (MeOH) λmax nm (log ε): 201 (3.30), 231 (3.33), 388 (1.75). For 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Table 1. HREIMS m/z: 210.1250 [M]+ (calcd for C12H18O3, 210.1256).
3.7 10-Lactyl vibralactone G (4)
Colorless oil; [α]−15.6(c 0.05, MeOH). UV (MeOH) λmax nm (log ε): 202 (3.74). IR (KBr) νmax cm−1: 3437, 2979, 2935, 2879, 1766, 1632, 1454, 1385, 1199, 1132, 1042, 956. For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Table 2. HRESIMS m/z: 279.1203 [M + Na]+ (calcd for C13H20O5Na, 279.1208).
3.8 (3S*,4R*)-6-Acetoxymethyl-2,2-dimethyl-3,4-dihydro-2H-chromene-3,4-diol (5)
Pale yellow solid; [α]−30.0 (c 0.02, MeOH). UV (MeOH) λmax nm (log ε): 203 (4.26), 230 (3.74), 280 (2.98). IR (KBr) νmax cm−1: 3451, 2924, 2935, 2871, 1723, 1639, 1497, 1384, 1252, 1033. For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Table 2. HREIMS m/z: 266.1147 [M]+ (calcd for C14H18O5, 266.1154).
3.9 Vibralactone Q (6)
Colorless oil; [α]−6.8 (c 0.09, MeOH). UV (MeOH) λmax nm (log ε): 204 (3.96). IR (KBr) νmax cm−1: 3427, 2972, 2932, 1718, 1640, 1450, 1403, 1383, 1200, 1134, 1063. For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Table 2. HREIMS m/z: 210.1259 [M]+ (calcd for C12H18O3, 210.1256).
References
M. Isaka, U. Srisanoh, W. Choowong, T. Boonpratuang, Org. Lett. 13, 4886–4889 (2011)
M. Isaka, U. Srisanoh, M. Sappan, S. Supothina, T. Boonpratuang, Phytochemistry 79, 116–120 (2012)
G.H. Li, F.F. Liu, L. Shen, H.J. Zhu, K.Q. Zhang, J. Nat. Prod. 74, 296–299 (2011)
J.C. Liermann, A. Schueffler, B. Wollinsky, J. Birnbacher, H. Kolshorn, T. Anke, T. Opatz, J. Org. Chem. 75, 2955–2961 (2010)
F.F. Liu, G.H. Li, Z.S. Yang, X. Zheng, Y. Yang, K.Q. Zhang, Helv. Chim. Acta 93, 1737–1741 (2010)
K. Ma, L. Bao, J.J. Han, T. Jin, X.L. Yang, F. Zhao, S.F. Li, F.H. Song, M.M. Liu, H.W. Liu, Food Chem. 143, 239–245 (2014)
X. Zheng, G.H. Li, M.J. Xie, X. Wang, R. Sun, H. Lu, K.Q. Zhang, Phytochemistry 86, 144–150 (2013)
D.Z. Liu, F. Wang, T.G. Liao, J.G. Tang, W. Steglich, H.J. Zhu, J.K. Liu, Org. Lett. 8, 5749–5752 (2006)
Q. Zhou, B.B. Snider, Org. Lett. 10, 1401–1404 (2008)
Q. Zhou, B.B. Snider, J. Org. Chem. 73, 8049–8056 (2008)
E. Zeiler, N. Braun, T. Böttcher, A. Kastenmüller, S. Weinkauf, S.A. Sieber, Angew. Chem. Int. Ed. 50, 11001–11004 (2011)
M.Y. Jiang, L. Zhang, Z.J. Dong, Z.L. Yang, Y. Leng, J.K. Liu, Chem. Pharm. Bull. 58, 113–116 (2010)
J.H. Ding, T. Feng, Z.H. Li, L. Li, J.K. Liu, Nat. Prod. Bioprospect. 2, 200–205 (2012)
G.Q. Wang, K. Wei, T. Feng, Z.H. Li, L. Zhang, Q.A. Wang, J.K. Liu, J. Asian Nat. Prod. Res. 14, 115–120 (2012)
G.Q. Wang, K. Wei, Z.H. Li, T. Feng, J.H. Ding, Q.A. Wang, J.K. Liu, J. Asian Nat. Prod. Res. 15, 950–955 (2013)
P.J. Zhao, Y.L. Yang, L. Du, J.K. Liu, Y. Zeng, Angew. Chem. Int. Ed. 52, 2298–2302 (2013)
L. Sun, D.L. Li, M.H. Tao, Y.C. Chen, Q.B. Zhang, F.J. Dan, W.M. Zhang, Nat. Prod. Res. 27, 1298–1304 (2013)
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (81102348).
Conflicts of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Chen, HP., Zhao, ZZ., Yin, RH. et al. Six New Vibralactone Derivatives from Cultures of the Fungus Boreostereum vibrans. Nat. Prod. Bioprospect. 4, 271–276 (2014). https://doi.org/10.1007/s13659-014-0029-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0029-z