
Abstract
Two new triterpenoids (1 and 2) and a new sterol (3), together with six known constituents (4–9), were isolated from the leaves and twigs of Melia azedarach. Their chemical structures were elucidated on the basis of spectroscopic analysis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Melia azedarach Linn. (Meliaceae) are widely distributed in southern districts of the Yellow River in China. The fruits and bark are commonly used as famous Traditional Chinese Medicine for acesodyne and disinsection [1]. This species has been reported to contain triterpenoids, steroids, limonoids, flavonoid glycosides, and simple phenolics [2], which have been found to possess some benefic pharmacological effects, including analgesic, anticancer, antiviral, antimalarial, antibacterial, and antifeedant activities [3, 4].
As a well known natural pesticide, azadirachtin has attracted much attention [5]. Previous investigations of the bark and roots of M. azedarach have shown that it is a rich source of meliacarpinin type limonoids [6–10]. Until now, few chemical studies have analyzed its leaves and twigs, which prompted us to conduct this project. We identified three new compounds: a meliacarpinin type limonoid (1), an apotirucallane derivative (2), and a sterol (3), together with six known compounds (4–9) (Fig. 1). Herein, we report the details of the isolation, structural elucidation of compounds 1–3.

The structures of compounds 1–9
2 Results and Discussion
The air-dried powder of M. azedarach leaves and twigs was extracted with MeOH (30 L × 3) at room temperature three times to give the residue, which was then partitioned between CHCl3 and water to get the CHCl3 soluble fraction. Then, three new constituents together with six known compounds were acquired by a series of chromatographic methods. Herein, we described the isolation and structural elucidation of these new compounds.
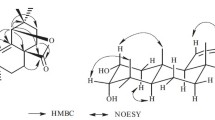
Compound 1 was isolated as an amorphous powder. The molecular formula was determined as C37H50O15 from the HREIMS ion peak at m/z 734.3159 [M]+ (calcd for 734.3150). Its IR spectrum showed the presence of hydroxyl (3456 cm−1) and carbonyl (1739 cm−1) groups. The 1D NMR data (Table 1) of 1 displayed characteristic signals of meliacarpinin skeleton with three methyls (δH 1.75, s, 3H; δH 0.95, s, 3H; δH 1.66, s, 3H), two methoxyls (δH 3.29, s, 3H; δH 3.79, s, 3H), two acetyls (δH 1.90, s, 3H; δH 2.30, s, 3H), one 2-methylbutyryl (δH 2.59, m; δH 1.27, d, J = 7.1 Hz; δH 1.53, m; δH 2.02, m; δH 0.99, t, J = 7.4 Hz) and one hydroxyl (δH 4.34, s, 1H) groups, which had a close resemblance to 3-tigloyl-1,20-diacetyl-11-methoxymeliacarpinin [8], except for the presence of one 2-methylbutyryl moiety in 1 instead of the tigloyl group at C-3 in. 3-tigloyl-1,20-acetyl-11-methoxymeliacarpinin. Observed the HMBC correlations (Fig. 2) of of H-2′ (δH 2.59, m), H-3′ (δH 1.27, d, J = 7.1 Hz), H-4′a (δH 1.53, m) with C-1′ (δC 176.1), and 1H-1H COSY correlations of H-3′/H-2′/H-4′/H-5′ (δH, 0.99, t, J = 7.4 Hz) confirmed above deduction. The linkage of 2-methylbutyryl moiety to C-3 was determined by the HMBC correlations from H-3 (δH 4.96, br. t, J = 2.7 Hz) to C-1 (δC 71.2), C-5 (δC 35.2), and C-1′.

Selected 1H-1H COSY ( ) and HMBC (
) and HMBC ( ) correlations of 1–3
) correlations of 1–3
The absolute configuration of C-2′ was determined as S, supported by the [α] 15D value at +16.3 of (S)-2-methylbutyric acid derived from 1 by alkaline hydrolysis ([α] 22D −14.3 for (R)-2-methylbutyric acid and [α] 25D +19.3 for (S)-2-methylbutyric acid) [11, 12]. The ROESY correlation (Fig. 3) between H-3 and H-6β (δH 4.12, br. d, J = 9.2 Hz) indicated that the 2-methylbutyryloxy was α-oriented. Other relative configuration of 1 were identical with those of 3-tigloyl-1,20-acetyl-11-methoxyneliacarpinin on the basis of ROESY spectrum. Therefore, chemical structure of 1 was deduced as 3α-(2-methylbutyryl)- 1,20-diacetyl-11-methoxymeliacarpinin.

Selected ROESY ( ) correlations of 1–3
) correlations of 1–3
Compound 2 was obtained as an amorphous powder. Based on the positive HREIMS (m/z 572.4083, calcd for 572.4077), the molecular formula was defined as C35H56O6. The 1H NMR, 13C-DEPT (Table 1) spectra showed the presence of nine methyls (two of which belonged to a tigloyl), eight methylenes (one oxygenated), eight methines (four oxygenated), one trisubstituted double bond, and four quaternary carbon. These data suggested that 2 was the apo-tirucallol (euphol) skeleton [13]. Comparison of NMR data of 2 with those of compound 5 (CAS NO: 1002345-41-6) revealed that they were similar [14], except that a senecioyl ester side chain at C-3 in compound 5 was replaced by a tigloyl group (δC 169.3 C-1′, 130.3 C-2′, 138.6 C-3′, 14.6 C-4′, and 12.4 C-5′) in 2 [8], which was confirmed by the HMBC correlations (Fig. 2) of H-3 (δH 4.65, t, J = 2.7 Hz), H-3′ (δH 6.92, qq, J = 7.1, 1.4 Hz), and H-5′ (δH 1.85, s-like) with C-1′, and of H-4′ (δH 1.81, dd, J = 7.1, 1.1 Hz) with C-2′, together with the 1H-1H COSY correlations of H-3′/H-4′.
The ROESY correlation (Fig. 3) between H-3 and Me-19β suggested that the tigloyl group at C-3 was α-oriented. The coupling constant between H-23 and H-24 (J = 9.0 Hz) suggested their anti-periplanar relation [14], and combination with the ROESY correlations of H-17/H-23, H-17/H-19β, H-20/Me-18α and H-24/Me-18α revealed that the configuration of C-23 and C-24 were both R*. Thus, the structure of 2 was established as 3α-tigloyl-17α-20S-21,24-epoxy-apotirucall-14-en-7α,23α,25-triol.
Compound 3 was isolated as an amorphous powder. The HREIMS of 3 gave a [M]+ ion peak at m/z 320.1985 (calcd for 320.1988), consistent with the molecular formula of C19H28O4. Detailed analysis of its 1H and 13C-DEPT (Table 2) and 2D NMR data indicated that 3 and 2α,3α-dihydroxyandrostan-16-one 2β,19-hemiketal [15] had the same planar structure. The only difference between them was the configuration of substituent group at C-3. Comparison its 1H NMR data with that of epi-isomer showed that the coupling constants of H-3 (δH 4.11, dd, J = 10.3, 6.0 Hz) and the chemical shifts for H-1α (δH 1.38, d, J = 11.3 Hz) and H-1β (δH 2.54, d, J = 11.3 Hz) were obviously different from those of 2α,3α-dihydroxyandrostan-16-one 2β,19-hemiketa. But the aforementioned data was familiar with 2α,3β-dihydroxypregnan-16-one 2β,19-hemiketal [10], which implied that the H-3 of 3 was α-oriented. This conclusion further confirmed by the cross peak between H-3 and H-5 (δH 1.38, overlap) in the ROESY spectrum (Fig. 3). So the hydroxyl group at C-3 was β-configuration. Consequently, the chemical structure of 3 was elucidated as 2α,3β-dihydroxyandrostan-16-one 2β,19-hemiketal.
Six known constituents: 1-cinnamoyl-3-acetyl-11-methoxymeliacarpinin (4) [8], 3-tigloyl-1,20-diacetyl-11-methoxymeliacarpinin (5) [8], 3S,23R,25-trihydroxytirucall-7-en-24-one (6) [16], and 2α,3α,16β-trihydroxy-5α-pregnane 20R-methacrylate (7) [17], 6-de(acetyloxy)-7-deacetylchisocheton compound E (8) [18], Toonapubesin C (9) [19], were identified by comparison of their spectroscopic data with those reported in the literature.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured with a Horiba SEPA-300 polarimeter. UV spectra were detected on a Shimadzu UV-2401A spectrophotometer. IR spectra were measured on a Bruker Tensor-27 infrared spectrophotometer with KBr pellets. ESIMS analysis were recorded on an API QSTAR Pulsar I spectrometer. EIMS and HREIMS were performed on a Waters Autospec Premier P776 mass spectrometer. 1D and 2D NMR spectra were recorded on Bruker DRX-500 and Bruker Avance III-600 spectrometers with TMS as internal standard. Semi-preparative HPLC studies were carried out on an Agilent 1100 liquid chromatograph with a Zorbax SB-C18 (9.4 mm × 25 cm) column. Column chromatography was performed with silica gel (200–300 mesh, Qingdao Marine Chemical, Inc.), Sephadex LH-20 (20–150 μm, Pharmacia), and Lichroprep RP-18 (40–63 μm, Merck). Fractions were monitored by TLC, and spots were visualized by heating the silica gel plates sprayed with 10 % H2SO4 in EtOH.
3.2 Plant Material
The leaves and twigs of M. azedarach were collected from Kunming, Yunnan Province, China. A voucher sample (NO: 2011-05-07) has been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried and powdered leaves and twigs of M. azedarach (10 kg) were extracted with MeOH (30 L × 3) at room temperature. Evaporation of the solvent under reduced pressure provide a dark residue (700 g), which was suspended in water and then partitioned with CHCl3 and n-BuOH, successively, to yield CHCl3 fraction (120 g), n-BuOH fraction (156 g). The CHCl3 extract was chromatographed by silica gel column eluted with CHCl3-MeOH as a gradient (100:1, 50:1, 20:1, 5:1) to afford four fractions. The CHCl3-MeOH (100:1) portion was evaporated to obtain a residue (20 g), which was subjected to silica gel chromatograph column with petroleum ether-EtOAc (10:1, 6:1, 3:1, 1:1) as elution, to give four fractions (A, B, C, and D). Fraction B (5 g) was further subjected to RP-18 chromatograph column, eluting with MeOH-H2O (40:60, 60:40, 80:20, and 100:0) to afford five fractions: B1–B5. Fraction B4 was then purified by HPLC (70 % CH3CN aq.; 2.0 mL/min; 210 nm; Zorbax SB-C18, 9.4 mm × 25 cm) to give compounds 1 (4 mg), 4 (2 mg) and 5 (3 mg). In the same way, 2 (4 mg), 6 (5 mg) and 9 (7 mg) were islated from fraction B3. Fraction B2 was subjected to silica gel chromatograph column with petroleum ether-EtOAc (8:1, 5:1, 3:1, 1:1, and 0:1) as elution, to give five subfractions (E, F, G, and H). Subfraction F was further separated and purified by silica gel chromatography column with CHCl3-Me2CO (50:1, 20:1, 5:1, and 1:1) as elution, get four subfraction: F1–F4, subfraction F2 was successively subjected to Sephadex LH-20 (MeOH) and HPLC (80 % CH3CN aq.; 2.0 mL/min; 210 nm; Zorbax SB-C18, 9.4 mm × 25 cm), and compounds 3 (1.5 mg), 7 (3 mg) and 8 (6 mg) were obtained.
3.4 3α-(2-Methylbutyryl)-1,20-diacetyl-11-methoxymeliacarpinin (1)
Amorphous powder; [α] 17D –17.8 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 208 (4.09) nm; IR (KBr) νmax 3456, 2953, 1739, 1706, 1618, 1438, 1376, 1252, 1160, 1131, 1061, and 949 cm−1; 1H NMR (500 MHz, C5D5N) and 13C DEPT (125 MHz, C5D5N) data, see Tables 1 and 2; positive ESIMS m/z 757 [M+Na]+; positive HREIMS m/z 734.3159 (calcd for C37H50O15 [M]+, 734.3150).
3.5 3α-Tigloyl-17α-20S-21,24-epoxy-apotirucall-14-en-7α,23α,25-triol (2)
Amorphous powder; [α] 17D –28.9 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 204 (3.80) nm; IR (KBr) νmax 3441, 2927, 2855, 1631, 1452, 1384, 1268, 1075 and 578 cm−1; 1H NMR (600 MHz, CD3OD) and 13C DEPT (150 MHz, CD3OD) data, see Tables 1 and 2; positive ESIMS m/z 595 [M+Na]+; positive HREIMS m/z 572.4083 (calcd for C35H56O6 [M]+, 572.4077).
3.6 2α,3β-Dihydroxyandrostan-16-one 2β,19-hemiketal (3)
Amorphous powder; [α] 17D –48.0 (c 0.30, MeOH); UV (MeOH) λmax (log ε) 202 (3.56), 219 (3.51) nm; IR (KBr) νmax 3464, 2924, 2874, 1720, 1447, 1295, 1187, 1130, 1044, and 993 cm−1; 1H NMR (600 MHz, C5D5N) and 13C DEPT (150 MHz, C5D5N) data, see Tables 1 and 2; positive ESIMS m/z 343 [M+Na]+; positive HREIMS m/z 320.1985 (calcd for C20H28O5 [M]+, 320.1988).
References
Editorial Committee of Flora of China, Chinese Academy of Science. In Flora of China, vol 43 (Science Press, Beijing, 1997), pp. 100–102
Z.S. Su, S.P. Yang, S. Zhang, L. Dong, J.M. Yue, Helv. Chim. Acta 94, 1515–1526 (2011)
S.B. Mahato, N.P. Sahu, G. Podder, Sci. Cult. 53, 29 (1987)
Vishnukanta, A.C. Rana, [Vishnukanta, 2008 #2350]
E.D. Morgan, Bioorg. Med. Chem. 17, 4096–4105 (2009)
M. Nakatani, R.C. Huang, H. Okamura, T. Iwagawa, Chem. Lett. 12, 2125–2128 (1993)
M. Nakatani, S. Arikawa, H. Okamura, T. Iwagawa, Heterocycles 38, 327–331 (1994)
K. Takeya, Z.S. Qiao, C. Hirobe, H. Itokawa, Phytochemistry 42, 709–712 (1996)
Q.G. Tan, X.N. Li, H. Chen, T. Feng, X.H. Cai, X.D. Luo, J. Nat. Prod. 73, 693–697 (2010)
F. Yoshiyasu, O. Mari, T. Hironobu, M. Hiroyuki, Chem. Pharm. Bull. 48, 301–303 (2000)
A.I. Meyers, G. Knaus, K. Kamata, J. Am. Chem. Soc. 96, 268–270 (1974)
B.B. Sylvia, B. George, M. George, J. Org. Chem. 32, 2641–2642 (1967)
X.D. Luo, S.H. Wu, D.G. Wu, Y.B. Ma, S.H. Qi, Tetrahedron 58, 6691–6695 (2002)
K. Mitsui, H. Saito, R. Yamamura, H. Fukaya, Y. Hitotsuyanagi, K. Takeya, Chem. Pharm. Bull. 55, 1442–1447 (2007)
M.T. Pupo, P.C. Vieira, J.B. Fernandes, M.F.G.F. da Silva, E. Rodrigues Filho, Phytochemistry 45, 1495–1500 (1997)
M. Arisawa, A. Fujita, N. Morita, P.J. Cox, R.A. Howie, G.A. Cordell, Phytochemistry 26, 3301–3303 (1987)
M. Nakatani, H. Takao, I. Miura, T. Hase, Phytochemistry 24, 1945–1948 (1985)
J. Li, M.Y. Li, T. Satyanandamurty, J. Wu, Helv. Chim. Acta 94, 1651–1656 (2011)
J.R. Wang, H.L. Liu, T. Kurtan, A. Mandi, S. Antus, J. Li, H.Y. Zhang, Y.W. Guo, Org. Biomol. Chem. 9, 7685–7696 (2011)
Acknowledgments
This work was supported financially by Joint Fund of NSFC and NSFY (No.U1132604), Key Program of MOST of CHINA (Nos. 2007BAD32B03 and SB2007FY400), as well as Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2010-ZZ14).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, WM., Liu, JQ., Peng, XR. et al. Triterpenoids and Sterols from the Leaves and Twigs of Melia azedarach. Nat. Prod. Bioprospect. 4, 157–162 (2014). https://doi.org/10.1007/s13659-014-0019-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0019-1