
Abstract
Staphylococcus aureus, a natural inhabitant of nasopharyngeal tract, survives mainly as biofilms. Previously we have observed that S. aureus ATCC 12600 grown under anaerobic conditions exhibited high rate of biofilm formation and l-lactate dehydrogenase activity. Thus, the concentration of pyruvate plays a critical role in S. aureus, which is primarily catalyzed by pyruvate kinase (PK). Analyses of the PK gene sequence (JN645815) revealed presence of PknB site in PK gene indicating that phosphorylation may be influencing the functioning of PK. To establish this hypothesis the pure enzymes of S. aureus ATCC 12600 were obtained by expressing these genes in PK 1 and PV 1 (JN695616) clones and passing the cytosolic fractions through nickel metal chelate column. The molecular weights of pure recombinant PK and PknB are 63 and 73 kDa, respectively. The enzyme kinetics of pure PK showed K M of 0.69 ± 0.02 µM, while the K M of PknB for stpks (stpks = NLCNIPCSALLSSDITASVNCAK) substrate was 0.720 ± 0.08 mM and 0.380 ± 0.07 mM for autophosphorylation. The phosphorylated PK exhibited 40 % reduced activity (PK = 0.2 ± 0.015 μM NADH/min/ml to P-PK = 0.12 ± 0.01 μM NADH/min/ml). Elevated synthesis of pyruvate kinase was observed in S. aureus ATCC 12600 grown in anaerobic conditions suggesting that the formed pyruvate is more utilized in the synthesis phase, supporting increased rate of biofilm formation.
Similar content being viewed by others

Introduction
Staphylococcus aureus mostly derives energy from glucose catabolism through glycolysis and Krebs cycles. The end product of glycolysis “Pyruvate” enters into TCA cycle and regulates the energy levels linked to pathogenicity of organism (Venkatesh et al. 2012). Pyruvate kinase (PK) belongs to a group of transferases that couples the free energy of PEP hydrolysis, using K+ and Mg+2 as co-factors where it generates ATP and pyruvate (Nowak and Suelter 1981). S. aureus generates two molecules of pyruvate for every molecule of glucose consumption that ultimately reduces two molecules of NAD+ to NADH creating redox imbalance which facilitates biofilm formation (Shimizu 2014; Ravcheev et al. 2012; Zhu et al. 2007).
“PK” is one of the three regulatory enzymes in glycolysis; it exhibited homotropic positive co-operativity for PEP, but not for ADP. It controls the entire glycolytic pathway by regulating the flux from Fructose-1, 6-bis-phosphate (FBP) to pyruvate (Muñoz and Ponce 2003). The most common form of allosteric regulation for PK is its upregulation by FBP, which increases the affinity and reduces the co-operativity of substrate binding which also depends on bound divalent cations in the active site. Here bound substrate and metal ions increases affinity of FBP for the allosteric site (Bond et al. 2000; Zoraghi et al. 2010, 2011a, b; Kumar et al. 2014). ATP, Alanine, and phenylalanine become negative allosteric inhibitors for PK and serves as a switch between the glycolytic and gluconeogenic pathways. This regulation flux by PK directly affects the concentrations of glycolytic intermediates, biosynthetic precursors, and nucleoside tri-phosphates in the cell. Thus PK controls consumption of metabolic carbon for biosynthesis and utilization of pyruvate for energy production (Shimizu 2014). Therefore; it appears that pyruvate levels are vital in the organism for the biofilm formation and maintaining of reductive conditions (Cramton et al. 1999; Gotz 2002; Yeswanth et al. 2013). In view of importance of pyruvate in this pathogen it was predicted that pyruvate kinase could be potential drug target and various studies using alkaloids as PK inhibitors were proposed as effective drugs against S. aureus infections (Zoraghi et al. 2010, 2011a, b); however, the essential role of pyruvate in the biofilm formation continues to pose questions against the efficacy of such compounds in vivo conditions.
The expression of enzymes involved in the cell wall biosynthesis, virulence factors, production of toxins, and purine biosynthesis for both energy production and growth are controlled through phosphorylation by PknB (Beltramini et al. 2009; De´barbouille et al. 2009; Donat et al. 2009; Tamber et al. 2010; Miller et al. 2010). The gene sequence of PK (JN645815) showed the presence of PknB site; therefore, like in Bacillus anthracis where fall in PK activity was observed on phosphorylation with PknB (Arora et al. 2012), we predicted that probably phosphorylation of PK might be controlling its function in S. aureus; hence the present study is aimed at understanding the effect of PK function on phosphorylation by PknB.
Materials and methods
In the present study chemicals were obtained from Sisco Research Laboratories Pvt. Ltd., India, Hi-Media Laboratories Pvt. Ltd., India, Sigma-Aldrich, USA, New England Biolabs, USA, and QIAGEN Inc., Valencia, CA, USA.
Bacterial strains and conditions
Staphylococcus aureus ATCC 12600 and Escherichia coli DH5α were obtained from Bangalore Genei Pvt Ltd. S. aureus was grown on modified Baird Parker agar at 37 °C. After overnight incubation, a single black shiny colony with distinct zone was picked and inoculated in Brain heart infusion (BHI) broth and incubated at 37° overnight. Thus grown S. aureus ATCC 12600 culture was used for the isolation of chromosomal DNA (Hari Prasad et al. 2012).
Pyruvate kinase enzyme assay
The pyruvate kinase enzyme assay was performed using crude and pure pyk (Venkatesh et al. 2012) and the kinetic parameters V max, K M, and K cat were calculated from Hanes-Woolf plot [S] vs ([S]/V). In all the experiments protein concentration was determined by the method of Bradford (1976).
Serine/Threonine protein kinase (PknB) assay
PknB activity was determined at 30 °C using novel nonradiolabeled protein kinase spectrophotometric assay with synthetic peptide acting as substrate on a Cyber lab spectrophotometer, USA. PknB assay mixture contained 0.1 M Tris–HCl pH 7.5, 0.1MATP, and 11.8 μM (30 µg/μl) peptide (stpks = NLCNIPCSALLSSDITASVNCAK). 1 µg/µl enzyme fraction (pure His tag PknB) was mixed and incubated at 30 °C for 10 min. The phosphorylated peptide was purified by passing it through Sephadex G-25 column (1 cm × 15 cm); the fractions were eluted with 0.1 M Tris–HCl pH 7.5 and 150 mM NaCl. The enzyme fraction appeared in the void volume, and in elution volume the phosphorylated peptide was obtained. The phosphate covalently bound to the proteins was estimated by adding freshly prepared reagent A (3.4 mM of Ammonium Molybdate dissolved in 0.5 mM H2SO4, 10 % SDS, 0.6 M l-Ascorbic acid mixed in 6:1:1(v/v/v) ratio) and incubated at 30 °C for 15 min and the absorbance was recorded at 820 nm against blank (0.1 M Tris–HCl pH 7.5 and 150 mM NaCl and reagent A (Clore et al. 2000). The enzyme activity was measured as the amount of phosphorous added per microgram peptide at 30 °C per minute per ml. For this, the calibration curve was developed using standard KH2PO4 for the estimation of inorganic phosphate and free phosphate was determined by adding reagent A (Fiske and Subbarow 1925). The phosphorylated peptide was further demonstrated by fractionating the eluted peptide on 15 % SDS-PAGE and staining the gel with reagent A; the bluish green-colored band that appeared in the gel indicated that peptide was phosphorylated by the enzyme fraction. Similarly, the auto-phosphorylation property of PknB was also determined and for this the reaction mixture composition was same except in that peptide was not added. The enzyme activity was measured as the amount of phosphorous added per microgram enzyme at 30 °C per minute per ml. Substrate level phosphorylation was performed by taking different substrate concentrations of 10–120 µM of synthetic peptide, keeping the ATP concentration constant, and the corresponding velocities were calculated and a graph of [S] vs [S/V] (Hanes-Woolf) was plotted, from the graph K M and V max was determined. For auto phosphorylation activity of PknB the same enzyme assay was carried out except in that peptide was not added. Similarly, the Km, V max for auto phosphorylation of PknB was determined by Hanes-Woolf plot.
Cloning of PknB and PK genes
PknB and PK genes were PCR amplified from chromosomal DNA of S. aureus ATCC 12600 and sequenced (Table 1); the amplified products were purified with NP-PCR Purification kit, Taurus Scientific, USA, and sequenced by dye terminating method at MWG Biotech India Ltd, Bangalore, India, and Xcelris Pvt. Ltd. Ahemadabad, India. Thus obtained gene sequences were deposited at GenBank (www.ncbi.nlm.nih.gov/genbank/submt.html) (Table 1) (Ohta et al. 2004). The PCR products were made into proper blunt ends using Klenow fragment (New England Bio labs, USA) and cloned in the Sma I site of pQE30 vector and transformed into E. coli DH5α, and generated clones were named as PV 1 and PK 1. The genes in clones PV 1 and PK 1 were over expressed with 0.75 mM IPTG and 1 mM IPTG for 2 and 5 h, respectively. The pure enzymes were obtained by passing the cytosolic fraction of each clone through nickel metal chelate column (Prasad et al. 2013). The expressed proteins were fractionated on 10 % SDS-PAGE and transferred on to nitrocellulose membrane (NCM) (Towbin et al.1979). The NCM was blocked with 2 % gelatin and treated with antiHis-tag monoclonal antibody kit (Qiagen) following the manufacturer’s method.
Multiple sequence alignments were carried out to understand the sequence similarities and dissimilarities. The PK sequences were retrieved for Staphylococcus aureus, Escherichia coli, Pyrobaculum aerophilum, Aeropyrum pernix, Streptococcus mutans, Bacillus licheniformis, Salmonella typhimurium, and Homo sapiens R/L isoform from GenBank. All these structures were subjected to CLUSTAL W software and the results were recorded (Thompson et al. 1997).
In vitro regulation of PK
PK was phosphorylated with PknB; the assay mixture contained 0.1 M Tris–HCl pH 7.5, 0.1 M ATP, 1 µg/ml pure enzyme (PK), and 3 µg/ml pure PknB was mixed and incubated at 30 °C for 10 min. The phosphorylated enzyme (PK) was purified by passing through Sephadex G-25 column (1 cm × 15 cm); the fractions were eluted with 0.1 M Tris–HCl pH 7.5 and 150 mM NaCl. The enzyme fractions appeared in the void volume. The bound phosphorous was estimated by adding freshly prepared reagent A and incubated at 30 °C for 15 min, and the absorbance was recorded at 820 nm against blank (0.1 M Tris–HCl pH 7.5 and 150 mM NaCl and reagent A). The phosphorylated enzymes were used to carry out the enzyme assay as described earlier. These phosphorylated proteins were fractionated in 10 % SDS-PAGE and the bound phosphate was detected by immersing the gel in reagent A and followed by staining with Coomassie Brilliant blue R250.
In all the experiments protein concentration was determined by the method of Bradford (1976).
Results
Cloning, expression, and characterization of PK gene
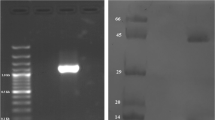
In the present study, the PK gene (1.7 kb) was PCR amplified from the chromosomal DNA of S. aureus ATCC 12600 and cloned in the Sma I site of pQE30 vector (Fig. 1a) in −1 frame, and the clone was named as PK 1. In order to ascertain the PK gene was sequenced (Supplementary Fig. 1) using the same primers and after ensuring the correct sequence (Accession number: JN645815), the enzyme was expressed in E. coli DH5α with 1 mM IPTG; thus expressed PK gene was purified by passing through nickel metal chelate agarose column. The pure recombinant PK exhibited single band in SDS-PAGE with a molecular weight of 63 kDa corresponding to the monomeric form of the enzyme (Fig. 1b) and the expression was validated using anti-His tag antibody (Fig. 1d). The PK sequence showed complete homology with all the PK gene sequence reported for other strains of S. aureus, indicating the presence of only one PK in this pathogen (Fig. 2). The rPK kinetics was close to the native PK (Table 2). The formation of pyruvate was higher in S. aureus grown in BHI broth compared to LB broth (Table 3).

a Electrophoretogram showing PCR amplification of pyruvate kinase from the chromosomal DNA of S. aureus ATCC 12600. Lane M super mix marker obtained from Merek Biosciences Pvt Ltd. L1 Amplified 1.7 kb PCR product. b SDS-PAGE analysis of analysis pyruvate kinase: electrophoretogram showing the pyruvate kinase protein obtained from recombinant clone PK 1. Lane M High-molecular-weight marker from Merek Biosciences Pvt Ltd. Lane 1 Cytosolic fraction of clone PK 1. Lane 2 Cytosolic fraction of clone PK 1 induced with IPTG. Lane 3 Purified PK obtained by passing the cytosolic fraction of IPTG-induced PK 1 clone through Nickel metal chelate agarose column. c In vitro phosphorylation assay: SDS-PAGE gel was first stained with reagent A, followed by Coomasie brilliant blue R250 staining. Lane M High-molecular-weight marker from Merek Biosciences Pvt Ltd. Lane L1 phosphorylated PK obtained from Sephadex G-25 column. Lane L2 Pure PK obtained from Nickel metal chelate agarose column. d Western blot using an anti-His tag antibody: Lane L1 phosphorylated PK obtained from Sephadex G-25 column. Lane L2 Pure PK obtained from Nickel metal chelate agarose column

Multiple sequence alignment of Pyruvate kinase: Amino acid sequences of Pyruvate kinase from various organisms were compared with the amino acid sequence of Human pyruvate kinase using CLUSTAL X
On scanning the annotated protein sequence of S. aureus PK in PROSITE (Altschul et al. 1997) the following exclusive sites were observed: casein kinase phosphorylation, N-myristoylation, N-glycosylation, protein kinase C phosphorylation, cAMP and cGMP-dependent protein kinase phosphorylation, cell attachment sequence, and Amidation sites which all indicate the pyk of S. aureus is a unique enzyme. The Multiple sequence alignment results showed very low amino acid sequence identity with other bacterial PK and human pyk (Fig. 2).
Cloning, expression, and characterization of PknB gene
The gene encoding PknB (2.0 kb) was amplified from S. aureus ATCC 12600 chromosomal DNA and sequenced; the sequence (JN695616) showed complete homology with PknB of several S. aureus strains in the reported databases. The sequence analysis of PknB enzyme showed the presence of catalytic domain between 10th and 267th residues which contains 12 specific Hank motifs, both ATP and substrate binding sites similar to eukaryotic protein kinases. Transmembrane domain followed by three different duplicated forms of PASTA domains. PASTA1 distributed between 377th and 440th residues, PASTA2 distributed between 445th and 508th residues, and PASTA3 distributed between 514th and 577th residues in the annotated protein sequence of PknB gene and between these two domains is a single transmembrane segment. These unique characters are the features exhibited by several PknB enzymes expressed in different strains of S. aureus.
The PknB gene was cloned in pQE 30vector and the clone was called as PV 1. PknB gene in this clone was expressed with 0.75 mM IPTG which resulted in the successful expression of the gene and the enzyme was purified by passing through nickel metal agarose column. The molecular weight of the rPknB was found to be 73 kDa which corresponds to the insert cloned and is equivalent to the monomeric form of Pkn B protein (Fig. 3a, b). The kinetics of PknB is explained in Table 4. The rPknB exhibited both substrate level phosphorylation and autophosphorylation; the phosphorylated molecules were separated on SDS-PAGE, and the appearance of blue colored bands on staining with reagent A confirms the presence of phosphate in the enzyme and substrate stpks (Fig. 3c).

a Electrophoretogram showing PCR amplification of PknB from the chromosomal DNA of S. aureus ATCC 12600. Lane M super mix marker obtained from Merek Biosciences Pvt Ltd. L1 Amplified 2 kb PCR product. b SDS-PAGE analysis of analysis PknB: electrophoretogram showing the PknB protein obtained from recombinant clone PV 1. Lane M High-molecular-weight marker from Merek Biosciences Pvt Ltd. Lane L1 Purified PknB obtained by passing the cytosolic fraction of IPTG-induced PV 1 clone through Nickel metal chelate agarose column. Lane L2 Cytosolic fraction of clone PV 1 induced with IPTG. Lane L3 Cytosolic fraction of clone PV 1. c In vitro phosphorylation assay: SDS-PAGE gel stained with reagent A. Lane L1 phosphorylated pure PknB obtained from Sephadex G-25 column. Lane L2 and L3 phosphorylated substrates of stpks obtained from Sephadex G-25 column
In vitro phosphorylation of PK
The presence of PknB site in the PK gene sequence encouraged us to carry out phosphorylation of PK. On phosphorylation with PknB, the bound phosphate was identified by its ability to react with reagent A which was identified spectrophotometrically. On fractionating these phosphorylated enzymes in 10 % SDS-PAGE and immersing the gel in reagent A, emergence of blue colored bands indicated PK was phosphorylated (Fig. 1c). The mobility of phosphorylated PK was higher than the native pure PK (Fig. 1c). The phosphorylated PK (P-PK) exhibited reduced activity (40 %) compared to the native PK (PK = 0.2 ± 0.015 μM NADH/min/ml to P-PK = 0.12 ± 0.01 μMNADH/min/ml) (Table 5).
Discussion
Staphylococcus aureus is a death-defying pathogen of both animals and humans that can cause minor skin infections to major life-threatening diseases. Phosphorylation of host proteins due to the secretary PknB has been implicated its ability to grow in any anatomical organs of human host (Lowy 1998; Miller et al. 2010). The expression of PknB in S. aureus is involved in controlling metabolic stress and regulation of plethora of metabolic pathways accordingly; we have found 40 % decreased PK activity on phosphorylation with PknB (Table 5). This response is upregulated in anaerobic growth conditions where Redox-sensing repressor Rex is reported to be involved in the regulation of anaerobic respiration in response to the NADH/NAD+ levels; in these conditions the association between pyruvate and TCA cycle is reported to be very weak, thus leading to more biosynthesis of toxins, virulence factors, and PIA synthesis which are highly favored for biofilm formation (Cramton et al. 1999; Beltramini et al. 2009; De´barbouille et al. 2009; Donat et al. 2009; Tamber et al. 2010; Liu et al. 2011; Strasters and Winkler 1963; Zhu et al. 2007; Pagels et al. 2010; Ravcheev et al. 2012).
In S. aureus, shift of growth conditions from aerobic to anaerobic increased the expression of glycolytic enzymes, such as GapA, Eno, Pgk, and Pyk (Fuchs et al. 2007); in our previous studies we have also shown elevated biofilm units and lactate dehydrogenase activity when S. aureus was grown in BHI broth with increased concentrations of glucose (Yeswanth et al. 2013). This enhanced glycolysis suppresses Krebs cycle resulting in the accumulation of lactate, acetate, formate, and acetoin, suggesting that glucose is catabolized to pyruvate further, catabolization via the lactate dehydrogenase, pyruvate formate-lyase, and butanediol pathway leading to biofilm formation (Zhu et al. 2007). In congruence with these observations in the present study, we have observed elevated PK activity in S. aureus grown in BHI broth compared to LB broth (Table 3). All these results conclusively explain the pyruvate formation in anaerobic condition favors more synthesis than energy generation contributing to the formation of biofilms which is one of the key pathogenic factors.
Conclusion
Staphylococcus aureus has unique feature to colonize on any anatomical locales of human body. This character makes the organism to spread its pathogenesis at a rapid rate. In this context, we found that PK catalyzes the irreversible conversion of Phosphoenolpyruvate to pyruvate that regulates the metabolic flux which is controlled by the expression of PknB. This PknB regulates functioning of PK thus controlling the levels of pyruvate in the organism. Therefore, high pyruvate formation in anaerobic conditions does not contribute to energy generation, but favors upregulating biosynthetic pathways involved in biofilm formation which is one of the key pathogenic factors.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Arora G, Sajid A, Arulanandh MD, Singhal A, Mattoo AR, Pomerantsev AP, Leppla SH, Maiti S, Singh Y (2012) Unveiling the novel dual specificity protein kinases in Bacillus anthracis: identification of the first prokaryotic dual specificity tyrosine phosphorylation-regulated kinase (DYRK)-like kinase. J Biol Chem 287(32):26749–26763
Beltramini AM, Mukhopadhyay CD, Pancholi V (2009) Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect Immun 77:1406–1416
Bond CJ, Jurica MS, Mesecar A, Stoddard BL (2000) Determinants of allosteric activation of yeast Pyk and identification of novel effectors using computational screening. Biochemistry 39:15333–15343
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Clore JN, Stillman J, Sugerman H (2000) Glucose-6-phosphatase flux in vitro is increased in type 2 diabetes. Diabetes 49:969–974
Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F (1999) The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67:5427–5433
De´barbouille M, Dramsi S, Dussurget O, Nahori MA, Vaganay E, Jouvion G, Cozzone A, Msadek T, Duclos B (2009) Characterization of a serine/threonine kinase involved in virulence of Staphylococcus aureus. J Bacteriol 191:4070–4081
Donat S, Streker K, Schirmeister T, Rakette S, Stehle T, Liebeke M, Lalk M, Ohlsen K (2009) Transcriptome and functional analysis of the eukaryotic-type serine/threonine kinase PknB in Staphylococcus aureus. J Bacteriol 191:4056–4069
Fiske CH, Subbarao Y (1925) The colorimetric determination of phosphorous. J Biol Chem 66:375–400
Fuchs S, Pané-Farré J, Kohler C, Hecker M, Engelmann S (2007) Anaerobic gene expression in Staphylococcus aureus. J Bacteriol 189(11):4275–4289
Gotz F (2002) Staphylococcus and biofilms. Mol Microbiol 43(6):1367–1378
Hari Prasad O, Nanda Kumar Y, Reddy OV, Chaudhary A, Sarma PVGK (2012) Cloning, expression, purification and characterization of UMP kinase from Staphylococcus aureus. Protein J 31(4):345–352
Kumar PS, Kumar YN, Prasad UV, Yeswanth S, Swarupa V, Sowjenya G, Venkatesh K, Srikanth L, Rao VK, Sarma PV (2014) In silico designing and molecular docking of a potent analog against Staphylococcus aureus porphobilinogen synthase. J Pharm Bioallied Sci 6(3):158–166
Liu Q, Fan J, Niu C, Wang D, Wang J, Wang X, Villaruz AE, Li M, Otto M, Gao Q (2011) The eukaryotic-type serine/threonine protein kinase Stk is required for biofilm formation and virulence in Staphylococcus epidermidis. PLoS One 6(9):e25380. doi:10.1371/journal.pone.0025380
Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339(8):520–532
Miller M, Donat S, Rakette S, Stehle T, Kouwen TRHM, Sander HD, Dreisbach A, Reilman E, Gronau K, Becher D, Peppelenbosch MP, Dijl JMV, Ohlsen K (2010) Staphylococcal PknB as the first prokaryotic representative of the proline-directed kinases. PLoS One 5(2):e9057. doi:10.1371/journal.pone.0009057
Muñoz ME, Ponce E (2003) Pyruvate kinase: current status of regulatory and functional properties. Comp Biochem Physiol B Biochem Mol Biol 135(2):197–218
Nowak T, Suelter C (1981) Pyk: Activation by and catalytic role of the monovalent and divalent cations. Mol Cell Biochem 35(2):65–75
Ohta T, Hirakawa H, Morikawa K, Maruyama A, Inose Y, Yamashita A, Oshima K, Kuroda M, Hattori M, Hiramatsu K, Kuhara S, Hayashi H (2004) Nucleotide substitutions in Staphylococcus aureus strains, Mu50, Mu3, and N315. DNA Res 11(1):51–56
Pagels M, Fuchs S, Pané-Farré J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Lalk M, Sander G, von Eiff C, Proctor RA, Engelmann S (2010) Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 76(5):1142–1161
Prasad UV, Vasu D, Kumar YN, Kumar PS, Yeswanth S, Swarupa V, Phaneendra BV, Chaudhary A, Sarma PV (2013) Cloning, expression and characterization of NADP-dependent isocitrate dehydrogenase from Staphylococcus aureus. Appl Biochem Biotechnol 169(3):862–869
Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL, Rodionov DA (2012) Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol 194(5):1145–1157
Shimizu K (2014) Regulation systems of bacteria such as Escherichia coli in response to nutrient limitation and environmental stresses. Metabolites 4:1–35
Strasters KC, Winkler KC (1963) Carbohydrate metabolism of Staphylococcus aureus. J Gen Microbiol 33:213–229
Tamber S, Schwartzman J, Cheung AL (2010) Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect Immun 78:3637–3646
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76(9):4350–4354
Venkatesh K, Srikanth L, Swarupa V et al (2012) Isolation, purification and characterization of Pyk from Staphylococcus aureus: a potential drug target. J Clin Sci Res 1:76–82
Yeswanth S, Nanda Kumar Y, Prasad UV, Swarupa V, Koteswara Rao V, Sarma PVGK (2013) Cloning and characterization of l-lactate dehydrogenase gene of Staphylococcus aureus. Anaerobe 24:43–48
Zhu Y, Weiss EC, Otto M, Fey PD, Smeltzer MS, Somerville GA (2007) Staphylococcus aureus biofilm metabolism and the influence of arginine on polysaccharide intercellular adhesin synthesis, biofilm formation, and pathogenesis. Infect Immun 75(9):4219–4226
Zoraghi R, See RH, Gong H, Lian T, Swayze R, Finlay BB, Brunham RC, McMaster WR, Reiner NE (2010) Functional analysis, overexpression, and kinetic characterization of pyruvate kinase from methicillin-resistant Staphylococcus aureus. Biochemistry 49:7733–7747
Zoraghi R, See RH, Axerio-Cilies P, Kumar NS, Gong H, Moreau A, Hsing M, Kaur S, Swayze RD, Worrall L, Amandoron E, Lian T, Jackson L, Jiang J, Thorson L, Labriere C, Foster L, Brunham RC, McMaster WR, Finlay BB, Strynadka NC, Cherkasov A, Young RN, Reiner NE (2011a) Identification of pyruvate kinase in methicillin-resistant Staphylococcus aureus as a novel, Antimicrobial Drug Target. Antimicrob Agents Chemother 55(5):2042–2053
Zoraghi R, Worrall L, See RH, Strangman W, Popplewell WL, Gong H, Samaai T, Swayze RD, Kaur S, Vuckovic M, Finlay BB, Brunham RC, McMaster WR, Davies-Coleman MT, Strynadka NC, Andersen RJ, Reiner NE (2011b) Methicillin-resistant Staphylococcus aureus (MRSA) pyruvate kinase as a target for bis-indole alkaloids with antibacterial activities. J Biol Chem 286(52):44716–447125
Acknowledgments
We sincerely acknowledge Sri Venkateswara Institute of Medical Sciences and University for providing facilities to carry out this work.
Conflict of interest
The authors declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Vasu, D., Sunitha, M.M., Srikanth, L. et al. In Staphylococcus aureus the regulation of pyruvate kinase activity by serine/threonine protein kinase favors biofilm formation. 3 Biotech 5, 505–512 (2015). https://doi.org/10.1007/s13205-014-0248-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13205-014-0248-3