
Abstract
An extensive literature review of the mechanistic modeling of n-heptane and cyclohexane pyrolysis was carried out. It was shown that Rice–Kossiakoff free radical theory does not adequately account for product distributions of n-heptane pyrolysis in the high conversion regime. Secondary reactions of alpha higher olefins and di-olefins accounted for the major products (ethene, propene and 1-butene) of n-heptane pyrolysis. Predicted product distributions (CH4, C2H4, C3H6, 1-C4H8 and 1,3-C4H6) of n-heptane pyrolysis showed very good agreement with experimental data. The product distributions of cyclohexane pyrolysis in the high conversion regime were rationalized and adequately accounted for using decomposition reactions of cyclohexyl bi-radicals followed by secondary reactions of major primary products such as C3H6 and 1,3-C4H6. The latter expanded mechanism can be used to model cyclohexane pyrolysis in the high conversion regime. Rate parameters (pre-exponential factors and activation energy) for each of the elementary reactions of n-heptane mechanistic model were either obtained from the literature or estimated using thermochemical parameters. The use of steady state approximation in mathematical modeling of n-heptane pyrolysis led to erroneous results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thermal decomposition of hydrocarbons is important in geothermal processes, conversion of petroleum oil, coal and biomass to liquid fuels, cracking of higher hydrocarbons to produce light olefins, degradation of endothermic jet fuels, and de-polymerization and recycling of synthetic polymers [1]. The pyrolysis of liquid and gaseous hydrocarbons is important industrially for the production of olefins, which are useful raw materials in the petrochemical industry. The products of the hydrocarbon pyrolysis reactions include ethylene, propylene and 1,3-butadiene. Most literature studies on hydrocarbons pyrolysis up to early 1980s were devoted to light paraffinic hydrocarbons because they result in high ethylene and propylene yields [2,3,4,5]. Secondly, majority of the work was done at sub atmospheric pressures, low temperatures and low conversions; the latter conditions are far removed from industrial practice at high conversion levels where secondary reactions are very significant. Thirdly, mechanisms were still not well understood. There was also a great need to investigate the thermal decomposition of other hydrocarbons because of the uncertainty of feedstock availability, which was at the time a major challenge facing olefins producers. In addition, expansion in the petrol-chemical industry, need for feedstock flexibility in pyrolysis plants, unstable prices and fierce competition among olefins manufacturers have provided the impetus for extensive research into the development of hydrocarbon pyrolysis models applicable to a wide range of feedstock from ethane and propane to whole crudes [6]. Hence, the design of new pyrolysis plants must incorporate feedstock flexibility from gaseous to liquid hydrocarbons. In the early 1980s there existed special need to investigate also liquid naphthenic feedstocks because petroleum crudes in Nigeria and a good number of countries as well as applicable crude fractions such as naphtha, are largely naphthenic. Furthermore, from the late 1980s to 2010 some researchers including the authors of this work reported on the pyrolysis of pure low and high molecular weight paraffins, olefins and naphthenes as well as mixtures of gaseous and liquid hydrocarbons under high conversion regime ([7,8,9,10,11,12,12,13,14,15,16,17,18]).
N-Heptane is an important component in many fuels and gives rise to high amount of heat energy when combusted [19]. In the range of alkanes from C5H12 to C12H26 n-heptane has the highest conversion and the highest yield of ethene at about 780 °C [20]. As a result many studies on hydrocarbon pyrolysis are devoted to n-heptane. Many studies have been reported on the thermal decomposition and catalytic pyrolysis of n-heptane in the past years [21,22,23,24].
The pyrolysis of heavy hydrocarbons is complex and the reaction mechanisms cannot be satisfactorily explained by the Rice–Kossiakoff (R–K) free radical theory [25, 26]. This is because the R–K Theory only accounts satisfactorily for the formation of products of the pyrolysis of light hydrocarbons at low temperatures and conversions (i.e. about 500–600 °C and 10%). The present authors suggested mechanistic models for the pyrolysis of pure low and high molecular weight hydrocarbons under conditions of high conversion [7,8,9,10].
Reliable quantitative kinetic models are required to design and optimize reactors for hydrocarbon pyrolysis. These models may be empirical or mechanistic (mechanism-based). Mechanistic models are better and, therefore, preferred over empirical ones as they give more information and deeper insight into the chemistry of hydrocarbon pyrolysis reactions. Secondly, they can predict the rate of reaction and the product distributions of the pyrolysis reactions under conditions beyond experimental range with greater confidence than empirical models [1]. However, mechanistic models require Arrhenius parameters for each elementary reaction in the mechanism as well as thermochemical properties such as enthalpy of formation, standard entropy, heat capacity for each chemical specie in the mechanism.
Better understanding of the mechanisms of the pyrolysis of hydrocarbons would be of great economic importance, when one considers the costs of petrochemical plants. Secondly, mechanistic models can be used to optimize thermal cracking operations and in reducing costly and time-consuming experimental programs [1, 27]. Mechanistic modeling is widely used thanks to the availability of fast computers. Though it is initially more expensive and time-consuming, its long term advantage is important; once it is developed, it is no longer necessary to employ an expensive pilot plant [28]. Furthermore, mechanistic models account extensively for free radical reactions, which play very significant roles in hydrocarbon pyrolysis. In addition, the pyrolysis of hydrocarbons under industrial conditions occurs at high conversions where secondary reactions of radicals predominate. Mechanistic models have been used mainly in the pyrolysis of hydrocarbons at low conversions [29,30,31]. There were few applications at high conversions before and in the early 1980s [5, 32, 33]. Also, most of the mechanistic modeling studies up to the late 1980s were on light hydrocarbons and there were few reported studies on the modeling of medium and heavy hydrocarbons pyrolysis [31, 34, 35].
From the late 1980s to date many studies have been reported on the mechanistic modeling of the pyrolysis of aliphatic, naphthenic and aromatic hydrocarbons [1, 7, 8, 9, 10, 36, 37, 38, 39, 40, 41]. Furthermore, criteria for the development of complex and non-regular kinetic schemes on hydrocarbon pyrolysis were reported in the literature [42]. Lumped parameter kinetic modeling of gaseous and liquid hydrocarbon pyrolysis, gasification and combustion reactions was reported [43, 44]. They lumped together large number of real components into properly selected number of equivalent components, thus facilitating the grouping of corresponding elementary reactions into equivalent or lumped reactions and the modeling of such reactions. Studies have been reported on coupled simulation of heat transfer and pyrolysis reactions in industrial cracking furnaces [45] and the development of an improved first principle-based mechanism that describes the molecular weight growth kinetics of ethane pyrolysis [46].
Studies have also been conducted on kinetic modeling of the formation of polycyclic aromatic hydrocarbons (PAHs) and soot during the pyrolysis of hydrocarbons as well as the thermal degradation of tar obtained from straw gasification and a computational fluid dynamics simulation of a reacting flow of partially combusting hot coke oven in a reformer [47,48,49,50,51,52].
In recent times investigations have been conducted on the mechanistic and kinetic modeling of the pyrolysis of hydrocarbons using Reactive Force Field (ReaxFF) Molecular Dynamics Simulation method [19, 53, 54]. This technique has been used to give atomic description of initiation mechanisms and product distributions of the pyrolysis and combustion of hydrocarbons as well as provide robust molecular reaction mechanisms required for kinetic modeling.
Hydrocarbons pyrolysis reactions may be described based on Free Radical theories of Rice–Kossiakoff [25, 26], Rice and Herzfeld [55], Fabuss-Smith-Satterfield [56] and Dente and Ranzi [27]. In modeling the thermal decomposition of hydrocarbons kinetic rate expressions are developed for radical and molecular species participating in the elementary reactions. The system of kinetic rate equations so developed are nonlinear first order ordinary differential equations (ODEs), which are stiff due to the wide disparity between the concentrations of molecular and radical species (10−2 and 10−10 M, respectively)—[31, 40, 57]. Consequently, they are complex and difficult to solve. Steady state approximation (SSA) is often postulated to simplify the complex mathematics associated with the numerical integration of stiff ODEs. Thus, SSA is employed to convert the ODEs representing radical species to system of non-linear algebraic equations. The SSA assumes that radical concentrations are constant during pyrolysis reactions, and this has been a subject of controversy in modeling hydrocarbon pyrolysis chemical reaction kinetic studies.
In view of the foregoing other workers have attempted to solve the system of ODEs without assuming SSA. Numerous techniques are available in the literature for the numerical integration of stiff ODEs [Gear’s DIFSUB 1971; LARKIN developed by [58]; basic semi-implicit Runge–Kutta method of Michelsen [59]; Adaptive semi-implicit Runge–Kutta (ASIRK) proposed by [60]; exponential collocation method of [61, 62]].
Research studies on hydrocarbons pyrolysis still continued as the design of pyrolysis plants based on laboratory data was not entirely successful. Also, minor technological improvements due, for instance, to a better understanding of the mechanisms of hydrocarbons pyrolysis would be of great economic importance. Furthermore, before 1970s most studies on the pyrolysis of hydrocarbons were carried out in batch or static reactors [63]; the use of open reactors commenced from that time, and their use is common in industrial practice till date.
The present study is aimed at reviewing the literature on kinetics and mechanistic modeling of the pyrolysis of pure hydrocarbons and their mixtures under high conversion regime. In doing this the present study will specifically focus on the pyrolysis of n-heptane and cyclohexane. The mechanistic models so developed will be validated using experimental data from our previous studies [7,8,9,10]. The study will provide insight into the kinetic modeling of the pyrolysis of high molecular weight hydrocarbons such as waste plastics and biomass as well as the combustion reactions of hydrocarbon fuels under high conversion regime.
Methodology
The experimental method and data used by the present authors to validate the proposed mechanisms for the thermal decomposition of n-Heptane and Cyclohexane in the high conversion regime have been presented elsewhere [7,8,9,10]. It is essentially a pulse kinetic set-up and it was adapted from the continuous-flow system used previously for the pyrolysis of cyclohexane [4, 5, 64]. Specifically, it comprised an annular tubular reactor made from 304 stainless steel, an electric furnace (Stanton Redcroft, UK), and three gas chromatographs, each with an integrating facility. Others were a liquid nitrogen cold trap and a split flow valve.
The annular reactor consisted of inner (i.d. 2.3 mm, o.d. 3.2 mm) and outer (i.d. 6.4 mm) tubes and had a surface-to-volume ratio (S/V) of 16 m cm−1. The temperature of the reactor was monitored in the same way as had been done in previous studies ([4, 5, 64].
The experiments were conducted in an annular tubular reactor at temperatures of 700–900 °C, space times of 0.40–1.14 s using the Pulse kinetic technique. Furthermore, experiments were conducted at 1 atmosphere pressure with excessive nitrogen gas dilution.
Mechanistic modeling studies
To develop mechanistic models, intrinsic reaction mechanisms for the pyrolysis reactions of the pure hydrocarbons investigated were determined based on the most plausible set of elementary reactions (ERs). In addition, the development of well-balanced mechanistic models is possible due to accurate and detailed thermo-chemical kinetic data and increased availability of pyrolysis data [1, 34, 36, 37, 40].
It is now generally accepted that hydrocarbons pyrolysis reactions may be described based on Free Radical theories of Rice–Kossiakoff [25, 26], Rice and Herzfeld [55], Fabuss-Smith-Satterfield [56] and Dente and Ranzi [27]. These chain reactions involve initiation, H-abstraction, addition, radical isomerization and decomposition elementary reactions. Others include disproportionation, aromatization and termination. It has been shown that the R–K theory is inadequate and cannot account for the product distributions of hydrocarbon pyrolysis in the high conversion regime [1, 7, 8, 9, 10, 34, 36, 40, 65]. Fabuss–Smith–Satterfield (F–S–S) involves the abstraction of hydrogen from the substrate by β radicals, which are formed by β-scission of the substrate-derived μ radicals. F–S–S chain theory has been shown to be more robust than the R–K theory [1, 40]. Another free radical theory of note is the Dente–Ranzi (D–R) theory, which has also been used extensively to develop mechanisms for hydrocarbon pyrolysis reactions [42, 44, 66]; D–R theory specifies the radical reaction mechanisms that explain the nature and type of reactions occurring in hydrocarbon pyrolysis in the high conversion regimes.
The chain mechanisms listed above were adapted in the present study to develop mechanisms for the pyrolysis of n-heptane and cyclohexane, which were used to generate kinetic rate expressions for radical and molecular species participating in the elementary reactions.
Consequently, elementary secondary radical reactions were included in the mechanism. Some molecular reactions were also included in the mechanistic models, since excluding them will result in erroneous results [67]. For instance, ethylene and 1,3-butadiene can undergo Diels–Alder concerted molecular reactions of the form shown below to form benzene as a secondary product in n-heptane pyrolysis:
These concerted molecular reactions can occur as fast as free radical reactions even with a smaller concentration of 1,3-butadiene [68]. The mechanisms for the thermal decomposition of n-heptane and cyclohexane under 1 atmosphere pressure and in the high conversion range are described as follows:
Mechanisms of n-heptane and cyclohexane pyrolysis
Selected key elementary reactions of n-heptane pyrolysis in the high conversion regime are described below. Detailed mechanistic models have been presented elsewhere [8].
Initiation
Hydrogen abstraction
Radical isomerization
Radical decomposition
To model n-heptane pyrolysis in the high conversion regime secondary reactions of alpha higher olefins such as 1-butene, 1-pentene and 1-hexene as well as di-olefins (1, 3-C4H6) should be included in the mechanism as they play very important roles in the pyrolysis reactions. The mechanistic model used for modeling n-heptane pyrolysis was adopted from an earlier model developed and presented elsewhere [7, 57].
Cyclohexane pyrolysis
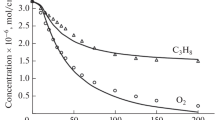
According Rice–Kossiakoff free radical theory cyclohexane undergoes initiation reactions to form bi-radicals, which undergo very fast isomerization reactions followed by the decomposition of the bi-radical isomers via the cleavage of the C–C bond β position relative to the unsaturated valence bond. A detailed description of the elementary reactions proposed to explain cyclohexane pyrolysis (Fig. 1) is presented elsewhere [4, 5, 69].

The H-abstraction reaction between C2H3 and C2H4 to form 1,3-C4H6 was reported by Norinaga and Deutschmann [50] to be important in pyrolysis reactions of olefins.
Thus, for an improved mechanistic modeling of cyclohexane pyrolysis secondary reactions of major primary products such as C3H6 and 1,3-C4H6 were proposed in this study.
In the high conversion regime some of the major primary products of cyclohexane pyrolysis, namely C3H6 and 1,3-C4H6 will undergo secondary reactions as explained in the case of n-heptane pyrolysis elsewhere [8]. Consequently, the secondary reactions in cyclohexane pyrolysis were expanded to include the following elementary reactions:
Decomposition (initiation) of olefins
Hydrogen exchange
Radical decomposition
Nomenclature
- 1-M-Al:
-
1-Methyl-allyl
- 3-BEN:
-
3-Butenyl
- 1-E-Al:
-
1-Ethyl-allyl
- 1-M-BEN:
-
1-Methyl-3-Butenyl
- 4-PEN:
-
4-Pentenyl
Selection of kinetic rate parameters
Kinetic rate parameters of the ER listed in the mechanistic model were compiled from several sources. Large numbers of accurate and reliable rate parameters of radical reactions of n-alkanes are now available in the literature [31, 33, 70, 71]. Kunugi et al. [72] also tabulated rate constants of free radical reactions of olefins. However, many of these parameters were compiled at low temperatures. The rate parameters of the pyrolysis reactions of C2–C4 alkanes at high conversions have been listed in the literature [33]. In this study, rate parameters of ER not available in literature were estimated using the generalized methods proposed by Benson [67]. A good number of other techniques have been outlined in the literature in the last 10–20 years [1, 38, 40, 50]. Thus, it has been shown that bond dissociation energies can be estimated to within − 1 to + 1 kcal/mol of the experimental values [38]. Dente–Ranzi Free Radical Theory on hydrocarbon pyrolysis reactions has also outlined the procedures for the estimation of kinetic rate parameters [42, 44, 66]. It is also pertinent to note that some of the rate parameters used in this study were modified within reasonable limits of − 2.5 to + 2.5% of their original values.
Mathematical models
To simulate the mechanistic models proposed for the pyrolysis of n-heptane and cyclohexane, systems of ordinary differential equations (ODE) were generated based on the continuity equations of the radical and molecular species participating in the pyrolysis reactions. The ODEs are given by:
and Sij is the stoichiometric constant for ith species in jth reaction. Furthermore, for the reactants Sij< 0 and Sij> 0 for products. A Computer Algorithm developed elsewhere [73] was used to translate the mechanistic models for n-heptane pyrolysis into mathematical models (sets of ODEs). Specifically, the Algorithm receives a mechanistic model as input and converts same to mathematical expressions of the form of Eqs. 2 and 3.
The ODEs were non-linear and “stiff” due to the wide disparity between the concentrations of molecular and radical species, 10−2 and 10−10 M, respectively [31, 40, 57]. Consequently, they are complex and difficult to solve. Steady state approximation (SSA) was employed to convert the ODEs representing radical species to system of non-linear algebraic equations shown below (Eq. 4):
The resulting system of nonlinear algebraic equations (Eq. 4) was then solved simultaneously using the Newton–Raphson technique, while the remaining ODEs were solved using the adaptive semi-implicit Runge–Kutta (ASIRK). On the other hand, the original set of ODEs was solved using LARKIN developed by Deuflhard et al. (1981). LARKIN is a numerical integration technique easily amenable to solving stiff ODEs.
Results and discussion
It is clear from Table 1 that percentage product mole fractions of ethylene, propylene, and 1-butene obtained in the n-heptane pyrolysis study were higher than predicted by R–K theory. Also the percentage product mole fractions reported for n-heptane pyrolysis [20, 35] were generally not in good agreement with the R–K theory. Thus, the R–K theory is not completely adequate in accounting for the product distributions of n-heptane pyrolysis in the high conversion regimes. These disparities may be rationalized in terms of the secondary decomposition reactions of the higher alpha olefins. Whereas the R–K theory predicts the formation of higher alpha olefins, particularly at low conversions, it fails to explain the secondary reactions they undergo to form lighter products including H2, CH4, C2H4 and C3H6. One set of such reactions is the H-abstractions of the α-olefins (1-C4H8, 1-C5H10, 1-C6H12) with small radicals (most likely CH3, C2H5 and less likely H and C3H5-allyl radicals [50, 74]. Such H-abstractions produce olefins, di-olefins and cyclo-olefins are shown below. Under high conversion conditions cited in the present study and used elsewhere [7,8,9,10] it is conceivable to propose that the cyclo-olefins will undergo dehydrogenation reactions to form aromatics during n-heptane pyrolysis. The presence of benzene as a secondary product supports this assertion.
Modeling studies
The mechanistic models of the thermal decomposition reactions of n-heptane were simulated with and without SSA. The predicted percentage mole fractions of the major products (CH4, C2H4, C3H6, 1-C4H8 and 1,3-C4H6) obtained for n-heptane pyrolysis using ASIRK (without SSA) showed a very good agreement with experiment data, while the results for simulation with SSA exhibited comparatively poor agreement with experimental data (Table 1).
In view of the foregoing, it was observed in earlier studies by the present authors [57] that SSA was clearly inadequate in the analysis of the reaction kinetics of hydrocarbon pyrolysis. It was confirmed elsewhere [57] that SSA was inadequate as associated simulated radical concentrations for n-heptane pyrolysis were observed to vary significantly with reaction time. These plots revealed that radical concentrations do not obey the SSA theory. They only appear to do so after the initial rapid increase. Whereas the primary radicals appeared to attain steady state, the secondary radicals did not exhibit this behavior. Secondly, the secondary radicals started to form when the primary radicals began to attain steady state. This implies that not all radicals appeared to attain steady state during n-heptane pyrolysis. Therefore, for simulation purposes radicals, which appear to attain steady state could be subjected to pseudo steady state approximation (PSSA), while others are treated the same way as molecular species.
This study was conducted in the high conversion regime because commercial pyrolysis plants normally operate in that range. Therefore, the results of this study are readily applicable to the design and operation of commercial plants.
A mechanism based on C–C bond scission was proposed elsewhere [8] to explain cyclohexane pyrolysis under high conversions. Hydrocarbons pyrolysis reactions are believed to occur through free radical mechanisms, based on Rice–Kossiakoff (R–K) free radical theory which involves chain processes, namely the initiation, propagation and termination steps. In our study the cleavage of the C–C bond at the beta position relative to the unsaturated valence bond was proposed as the initiation step in the pyrolysis of cyclohexane, leading to the formation of cyclohexyl bi-radicals (Fig. 1). This step was followed subsequently by fast isomerization reactions of leading to the formation of the olefins (ethene, propene, and 1,3-butadiene) which were identified experimentally as the major products of cyclohexane pyrolysis.
It is conceivable that elementary reactions proposed in “Mechanisms of n-heptane and cyclohexane pyrolysis” can be used to develop mechanistic models applicable for modeling cyclohexane pyrolysis in the high conversion regime.
Inadequacy of steady state approximation (SSA) in modeling hydrocarbon pyrolysis
Modeling allows confident extrapolation outside the range of data used in its development. Modeling can be used in optimizing chemical processes and operations and in reducing costly and time-consuming experimental programs.
The mechanistic models of the thermal decomposition reactions of n-heptane were simulated with and without SSA. The predicted product distributions obtained for n-heptane pyrolysis using ASIRK (without SSA) showed a very good agreement with experimental data, while the data predicted using the model based on SSA exhibited comparatively poor agreement with experimental data. It is, therefore, conceivable to note that SSA is clearly inadequate in rationalizing the reaction kinetics of hydrocarbon pyrolysis. Our study confirmed that the application of SSA in kinetic analysis of hydrocarbons pyrolysis reactions should be discontinued.
References
Savage PE (2000) Mechanisms and kinetic models for hydrocarbons pyrolysis. J Anal Appl Pyrolysis 54(1–2):109–126
McConnell CF, Head BD (1983) Chapter 12. In: Albright LF, Crynes BL, Corcoran WH (eds) Pyrolysis: theory and industrial practice. Academic, New York
Corcoran WH (1983) Chapter 3. In: Albright LF, Crynes BL, Corcoran WH (eds) Pyrolysis: theory and industrial practice. Academic, New York
Aribike DS, Susu AA, Ogunye AF (1981) Kinetics of the thermal decomposition of cyclohexane. Thermochim Acta 47:1–14
Aribike DS, Susu AA, Ogunye AF (1981) Mechanistic and mathematical modeling of the thermal decomposition of cyclohexane. Thermochim Acta 51:113–127
Hirsig AR, Schlanger MO (1984) How can the olefins industry survive in the 80s? Chem Eng Prog 80(2):24
Aribike DS, Susu AA (1988) Kinetics and mechanism of the thermal cracking of n-heptane. Thermochim Acta 127:247–258
Aribike DS, Susu AA (1988) Kinetics of the pyrolysis of cyclohexane using the pulse technique. Ind Eng Chem Res 27:915–920
Aribike DS, Susu AA (1988) Thermal cracking of n-butane and a light hydrocarbon mixture. J Anal Appl Pyrol 14:37–48
Aribike DS, Susu AA (1988) Mechanistic modeling of the pyrolysis of n-heptane. Thermochim Acta 127:259–273
Zhao Y, Zhang S, Li D (2014) Understanding the mechanism of radical reactions in 1-hexene pyrolysis. Chem Eng Res Des 92(3):453–460
Laufer AH, Fahr A (2004) Reaction and kinetics of unsaturated C2 hydrocarbon radicals. Chem Rev 104(6):2813–2832
Aribike DS, Susu AA (2006) Autocatalysis in n-heptane pyrolysis. J Sci Tech Environ 6(1&2):83–92
Behar F, Lorant F, Budzinski H, Desavis E (2002) Thermal stability of alkyl aromatics in natural systems: kinetics of thermal decomposition of dodecylbenzene. Energy Fuels 16:831
Wolf RA, Trocino RJ, Rozich WR, Sabeta IC, Ordway RJ (1998) Thermal decomposition of tert-butyl 1 arylcycloalkane percarboxylates. J Org Chem 63:3814
Murata M, Takeda N, Saito S (1974) Simulation of pyrolysis of paraffinic hydrocarbon binary mixtures. J Chem Eng Jpn 7:286
Yu J, Eser S (1997) Thermal decomposition of C10–C14 normal alkanes in near-critical and supercritical regions: product distributions and reaction mechanism. Ind Eng Chem Res 36:585
Yu J, Eser S (1998) Thermal decomposition of jet fuel model compounds under near-critical and supercritical conditions. 2. Decalin and tetralin. Ind Eng Chem Res 37:4601
Li J, Wang F, Cheng X, Li X (2013) Reactive molecular dynamics simulation on thermal decomposition of n-heptane. Chin J Chem Phys 26(2):211–219
Bajus M, Vesely V, Leclercq PA, Rijks JA (1979) Steam cracking of hydrocarbons. I. Pyrolysis of heptane. Ind Eng Chem Prod Res Dev 18:30
Watson BA, Klein MT, Harding RH (1996) Mechanistic modeling of n-heptane cracking on HZSM-5. Ind Eng Chem Res35:1506
Pant KK, Kunzru D (1997) Catalytic pyrolysis of n-heptane: kinetics and modeling. Ing Eng Chem Res. 36:2059
McEnally CS, Ciuparu Pfefferle LD (2003) Experimental study of fuel decomposition and hydrocarbon growth processes for practical fuel components: Heptanes. Combust Flame 134:339
Ding JX, Zhang L, Han KL (2011) Thermal rate constants of the pyrolysis of n-heptane. Combust Flame 158(12):2314–2324
Kossiakoff A, Rice FO (1943) Thermal decomposition of hydrocarbons resonance stabilization and Isomerization of Free Radicals. J Am Chem Soc 65:590
Rice FO (1931) The thermal decomposition of organic compounds from the standpoint of free radicals. I. Saturated hydrocarbons. J Am Chem Soc 53:1959
Dente ME, Ranzi EM (1983) In: Albright LF, Crynes BL, Corcoran WH (eds) Pyrolysis: theory and industrial practice. Academic, New York, p 133
Ross LL, Shu WR (1979) In: Oblad AG, Davis HG, Eddinger RT (eds) Thermal hydrocarbon chemistry, Adv. Chem. Ser., vol 183. American Chemical Society, Washington, p 129
Pacey PD, Purnell JH (1972) Propylene from paraffin pyrolysis. Ind Eng Chem Fundam 11:233
Powers DR, Corcoran WH (1974) Pyrolysis of n-butane – explicit effects of primary and secondary butyl radicals and of secondary reactions. Ind Eng Chem Fundam 13:351
Isbarn GHJ, Ederer HJ, Ebert KH (1981) In: Ebert KH, Deuflhard P, Jaeger W (eds) Modeling of chemical reaction systems; springer series in chem. phys, vol 18. Springer, New York, p 235
Layokun SK, Slater DH (1979) Mechanism and kinetics of propane pyrolysis. Ind Eng Chem Process Des Dev 18(2):232
Sundaram KM, Froment GF (1978) Modeling of thermal cracking kinetics: 3. Radical mechanisms for the pyrolysis of simple paraffins, olefins and their mixtures. Ind Eng Chem Fundam 17:174
Ranzi EM, Dente M, Pierucci S, Biardi G (1983) Initial product distributions from pyrolysis of normal and branched paraffins. Ind Eng Chem Fundam 22:132
Murata M, Saito S, Amano S, Maeda S (1973) Prediction of initial product distributions from pyrolysis of normal paraffinic hydrocarbons. J Chem Eng Jpn 6:252
Jess A (1996) Mechanisms and kinetics of thermal reactions of aromatics hydrocarbons from pyrolysis of solid fuels. Fuel 75(12):1441–1448
Norinaga K, Sakurai Y, Sato R, Hayashi J (2011) Numerical simulation of thermal conversion of aromatic hydrocarbons in the presence of hydrogen and steam using a detailed chemical kinetic model. Chem. Eng. J. 178:282–290
Xiao Y, Longo JM, Hieshima GB, Hill RJ (1997) Understanding the kinetics and mechanisms of hydrocarbon thermal cracking: an ab initio approach. Ind Eng Chem Res 36:4033–4040
Burkle-Vitzthum V, Bounaceur R, Marquaire PM, Montel F, Fusetti L (2011) Thermal evolution of n-, and iso-alkanes in oils. Part 1: pyrolysis model for a mixture of 78 alkanes (C1–C32) including 13,206 free radicals reactions. Org Geochem 42:439–450
Savage PE (1990) Pyrolysis of a binary mixture of complex hydrocarbons: reaction modeling. Chem Eng Sci 45(4):859–873
Poutsma ML (2000) Fundamental reactions of free radicals relevant to pyrolysis reactions. J Anal Appl Pyrolysis 54:5–35
Dente M, Pierucci S, Ranzi E, Bussain G (1992) New improvements in modeling kinetic schemes for hydrocarbon pyrolysis reactors. Chem Eng Sci 47:2629–2634
Ranzi E, Dente M, Goldaniga A, Bozzano G, Faravelli T (2001) Lumping procedures in detailed kinetic modeling of gasification, pyrolysis, partial oxidation and combustion of hydrocarbon mixtures. Prog Energy Combust Sci 27:99–139
Pierucci S, Ranzi S, Dente M, Barendregt S (2005) In: Puigjaner L, Espuna A (eds) A Kinetic generator of hydrocarbon pyrolysis mechanisms. European Symposium on Computer-Aided Process Engineering-15
Niaei A, Towfighi J, Sadrameli SM, Karimzadeh R (2004) Combined simulation of heat transfer and pyrolysis reactions in industrial cracking furnaces. Appl Therm Eng 24:2251–2265
Xu C, Al-Shoaihi AS, Wang C, Carstensen HH, Dean AM (2011) Kinetic modelling of ethane pyrolysis at high conversions. J Phys Chem A 115(38):10470–10490
Saggese C, Sanchez NE, Frassoldati A, Cuoci A, Faravelli T, Alzueta MU, Ranzi E (2014) Kinetic modeling study of polycyclic aromatic hydrocarbons (PAHs) and soot formation in acetylene pyrolysis. Energy Fuels 28(2):1489–1501
Ranzi E, Frassoldati A, Granata S, Faravelli T (2005) Wide-range kinetic modeling study of the pyrolysis, partial oxidation and combustion of heavy n-alkanes. Ind Eng Chem Res 44(14):5170–5183
Djokic MR, Van Geem KM, Cavallotti C, Frassoldati A, Ranzi E, Marin GB (2014) An experimental and kinetic modeling study of cyclopentadiene pyrolysis: first growth of polycyclic aromatic hydrocarbons (PAHs). Combust Flame 161(11):2739–2751
Norinaga K, Deutschmann O (2007) Detailed kinetic modeling of gas-phase reactions in the chemical vapor deposition of carbon from light hydrocarbons. Ind Eng Chem Res 46:3547–3557
Chen G, Liu C, Yan B, Ma W (2014) Thermal degradation behaviours and kinetics of biomass tar. Energy Procedia 61:1085–1088
Li C, Appari S, Tanaka R, Hanao K, Lee Y, Kudo S, Hayashi J, Janardhanan VM, Watanaje H, Norinaga K (2015) A CFD study on the reacting flow of partially combusting hot coke oven gas in a bench-scale reformer. Fuel 159:590–598
Wang Q, Wang J, Li J, Tan N, Li X (2011) Reactive molecular dynamics simulation and chemical kinetic modeling of pyrolysis and combustion of n-dodecane. Combust Flame 158(2):217–226
Van Duin ACT, Dasgupta S, Lorant F, Goddard WA III (2001). ReaxFF: A reactive force field for hydrocarbons. J Phys Chem A 105:9396
Rice FO, Herzfeld KF (1934) The thermal decomposition of organic compounds from the standpoint of free radicals. VI. The mechanism of some chain reactions. J Am Chem Soc 56:284
Fabuss BM, Smith JO, Satterfield CN Jr (1964) In: McKetta JJ Jr (ed) Advances in petroleum chemistry and refining, vol 9. Wiley, New York, p 179
Aribike DS, Susu AA (1988d) Copyrolysis of n-heptane and cyclohexane in the presence of benzene. In: Refereed proceedings of the Nigerian Society of Chemical Engineers annual conference and general meeting held at the Petroleum Training Institute, Warri, Nigeria, Nov 9–12, pp 69–79
Deuflhard P, Bader G, Nowak U (1981) LARKIN – a software package for the numerical simulation of LARge systems arising in chemical reaction KINetics. In: Ebert K, Deuflhard P, Jaeger W (eds) Modeling of chemical reaction systems. Springer Series in Chem Phys, vol 8. Springer New York, p 38
Mchelsen ML (1976) An efficient general purpose method for the integration of stiff ordinary differential equations. AICHE J 22:594
Prokopakis GJ, Seider WD (1981) Adaptive semi-implicit Runge-Kutta method for solution of stiff ordinary differential equations. Ind Eng Chem Fundam. 20:255
Guertin EW, Sorensen JP, Stewart WE (1977) Exponential collocation of stiff reactor models. Comput Chem Eng 1:197
Aribike DS, Agunu BO, Olafadehan OA (2006) On steady state approximation in modeling n-hexane and n-heptane pyrolysis reactions. J Sci Tech Environ 6(1&2):52–63
Come GM. In: Albright LF, Crynes BL, Corcoran WH (eds) (1983) Pyrolysis: theory and industrial practice. Academic Press, New York, p 275
Susu AA, Ogunye AF (1978) Selective naphthene pyrolysis for ethylene with hydrogen as diluent. Thermochimica Acta 34:197
Blouri B, Giraud J, Nouri S, Herault D (1981) Steam cracking of high-molecular weight hydrocarbons. Ing Eng Chem Process Des Dev 20:307–313
Ranzi E, Faravelli T, Gaffuri P, Garavaglia E,Goldaniga A (1997) Primary pyrolysis and oxidation reactions of linear and branched alkanes. Ind Eng Chem Res 36(8):3336
Benson SW (1978) Thermochemical kinetics, 2nd edn. Wiley, New York
Sakai T (1976) Thermal reaction of olefins and diolefins, and formation of aromatics. In: Albright LF, Crynes BL (eds) Industrial and laboratory pyrolysis. American Chemical Society Symposium Series, No 32, p 89. ACS, Washington DC
Aribike DS (1986) Comparative kinetic investigation of the pyrolysis of pure hydrocarbons and their mixtures. PhD Thesis, University of Lagos, Lagos, Nigeria
Trotmann-Dickenson AF (1965) Advanced free-radical chemistry, vol 1. Wiley, New York
Kerr JA (1976) In: Bamford CH, Tipper CFH (eds) Comprehensive chemical kinetics, vol 18, p 39. Elsevier, Amsterdam
Kunugi T, Sakai T, Somak Sasaki Y (1969) Kinetics and mechanism of thermal reaction of ethylene. Ind Eng Chem Fundam 8:374
Aribike DS, Agunu BO (2007) Modeling of thermal decomposition reactions of hydrocarbons: development of a computer algorithm. J Sci Tech Environ 7(1&2):1–7
Rebick C (1976) Pyrolysis of heavy hydrocarbons. In: Albright LF, Crynes BL (eds) Industrial and laboratory pyrolysis. American Chemical Society Symposium Series, No 32, p 69. ACS, Washington DC
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aribike, D.S., Susu, A.A. A review of mechanistic and mathematical modeling of n-heptane and cyclohexane pyrolysis. Appl Petrochem Res 8, 193–201 (2018). https://doi.org/10.1007/s13203-018-0213-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-018-0213-x