
Abstract
In a natural analog study of risks associated with carbon sequestration, impacts of CO2 on shallow groundwater quality have been measured in a sandstone aquifer in New Mexico, USA. Despite relatively high levels of dissolved CO2, originating from depth and producing geysering at one well, pH depression and consequent trace element mobility are relatively minor effects due to the buffering capacity of the aquifer. However, local contamination due to influx of brackish waters in a subset of wells is significant. Geochemical modeling of major ion concentrations suggests that high alkalinity and carbonate mineral dissolution buffers pH changes due to CO2 influx. Analysis of trends in dissolved trace elements, chloride, and CO2 reveal no evidence of in situ trace element mobilization. There is clear evidence, however, that As, U, and Pb are locally co-transported into the aquifer with CO2-rich brackish water. This study illustrates the role that local geochemical conditions will play in determining the effectiveness of monitoring strategies for CO2 leakage. For example, if buffering is significant, pH monitoring may not effectively detect CO2 leakage. This study also highlights potential complications that CO2 carrier fluids, such as brackish waters, pose in monitoring impacts of geologic sequestration.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There is understandable concern about possible impacts to shallow aquifers if CO2 were to leak from primary storage reservoirs during or after geologic sequestration operations. There is a paucity of field data, however, to characterize the nature of potential impacts. Undesirable side affects might include catastrophic release of CO2 at the surface (e.g., well blow-out) and leakage of brine (Benson 2002; Kharaka et al. 2006). Although CO2 is not toxic in low-concentrations, dissolution into shallow groundwater may depress pH and subsequent trace metal dissolution from aquifer minerals may increase the concentration of naturally-occurring toxic elements such as lead and arsenic (Benson 2002; Wang and Jaffe 2004). Predicting the potential impact of CO2 release in shallow aquifers at any sequestration site is a challenging task. There are at least three possible research strategies for evaluating this type of effect, all of which have strengths and limitations. One is to directly monitor shallow aquifer chemistry at engineered CO2 sequestration sites where CO2 is leaking upward at a known rate. This may not be feasible because engineered sites are generally thought NOT to be leaking, by design, and so directly measuring the impacts of leaks is difficult. Even if leaks do occur, few sites have background groundwater monitoring programs in place that would allow quantitative comparison of pre- and post-leak water quality (Liewicki et al. 2007). Leaks may be difficult to detect due to limitations in infrastructure, low density of wells, and infrequent measurements. Consequently, establishing robust monitoring strategies and characterizing background geochemical conditions has become a high priority at some engineered sites (Smyth et al. 2006). Another strategy involves using mineralogy and water quality data collected from a variety of aquifer types that might ultimately receive CO2 leaks and predict possible impacts using geochemical theory (Birkholzer et al. 2007; Birkholzer 2008). This strategy can provide insight into processes that may ultimately be important. However, the predictive ability of studies like these will always be limited by irreducible uncertainty in field-scale mineral dissolution kinetic rates and local-scale heterogeneity in aquifer mineralogy. Mechanistic-based approaches to measuring kinetics are only possible in the laboratory under well-controlled conditions (Bose and Sharma 2002; Bruno et al. 1992; Craw et al. 2003; Golubev et al. 2005; Stephens and Hering 2004). Unfortunately, large discrepancies between field and laboratory rate estimates (Chen 2005; Langmuir 1997) contributes to uncertainty in predictive modeling. The impact of uncertainty in kinetic rate on predicting trace metal mobility due to CO2 leaks has been well documented (Wang and Jaffe 2004). Another factor which will limit the predictive ability of geochemical models is the difficulty of predicting scavenging of trace metals by secondary minerals such as carbonates, iron oxy-hydroxides and clay minerals (Aiuppa et al. 2005). Without validation at natural or engineered analog sites where CO2 is present at significant concentrations, it will be difficult to assess the accuracy of this modeling-based approach.
A third approach, utilized in this study, involves directly measuring CO2 impacts at a natural analog site where CO2 is actively upwelling through a shallow aquifer. These sites are usually located in volcanic or geothermal settings (Federico et al. 2004; Glennon 2005; Lu et al. 2005). Like the other two strategies, this empirical approach has inherent limitations. Most importantly, the highly fractured geologic settings where active CO2 upwelling occurs naturally are very unlikely to be considered for geologic sequestration (Benson 2002). In addition, the lack of “pre CO2 flux” water quality data inhibits comparison of pre- and post-CO2 conditions. The aquifer may not be impacted only by CO2, but rather a mixture of CO2 and deep thermal waters of unknown origin and/or composition. A dense network of wells available for shallow groundwater characterization may not be available. With these caveats in mind, field observations and interpretations of a site in northern New Mexico with active CO2 upwelling through a shallow aquifer are presented here. A number of domestic wells are available for sampling, offering an opportunity to compare background groundwater quality and CO2-impacted groundwater quality. Conclusions follow, with implications for CO2 sequestration leakage scenarios and monitoring strategies.
Natural analog sites for studying CO2 impacts on aquifers
There are two major categories of natural analogs: (1) locations where diffuse CO2 is rising and flowing through an aquifer (where it can be measured in wells), and (2) locations where CO2 is rising along a fault or other conduit and expresses itself at a point at the ground surface (a spring or a geyser) (Fig. 1). Studies of the second type are far more common, due in part to the ease of sampling springs or geysers. Studies of CO2-charged springs (Glennon 2005; Goff et al. 1988; Newell et al. 2005; Vuataz et al. 1984) provide insight into the dynamics of CO2 flow and into geochemical processes occurring at depth, but they are not particularly informative about the impact of CO2 on aquifers, since the CO2 may not significantly impact a shallow aquifer if it rises along a conduit and rapidly discharges at a spring (Fig. 1a). However, the extent that CO2 upwelling along faults mixes with local groundwater may be assessed by temperature and trace elements (Vuataz et al. 1984).

Two types of natural analogs (a) CO2 rises with deep water along a fault and forms a CO2-rich spring, b CO2 rises with deep water along a faults and diffuses into shallow aquifer water. CO2 degasses at springs and also along the water table to the vadose zone
Far less common are studies of aquifer chemistry in CO2 rich settings that include samples from both wells and springs. Very comprehensive studies on groundwater chemistry in high CO2 flux settings have been performed on volcanoes in Italy (Aiuppa et al. 2005; Federico et al. 2004). Here, large datasets on groundwater chemistry in aquifers (including but not limited to springs) have been collected and analyzed using very sophisticated geochemical modeling. The authors were able to discriminate between trace metal concentrations controlled by availability in aquifer materials (As, Cd, Mo, Se, V) and trace metal concentrations kinetically-controlled in mineral dissolution/precipitation. Because of dissolution/desorption of trace metals from aquifer minerals and subsequent scavenging by secondary minerals (primarily calcite), no trends in trace element concentrations with \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) or pH (range 5.8–8.0) were measured. Mammoth Mountain, CA (Rogie et al. 2001) also represents a diffuse CO2 flux volcanic setting. Here, CO2 flux resulted in localized tree mortality. Elevated trace metal concentrations and depressed pH (near 4.0) were measured in the soil profile; however, geochemical data measured in springs and wells did not show a clear relation between elevated trace metal concentrations in groundwater with either pH (5.1–8.46) or \( {\text{P}}_{{{\text{CO}}_{ 2} }}.\) One conclusion drawn by the authors was that the effect of upwelling CO2 is “substantially neutralized” by mineral weathering, which in turn increases the total dissolved solids (TDS) of the fluid (Evans et al. 2002).
The site described in this study not only has a CO2 expression at the ground surface (e.g., eruptions of a cold geyser) but also in a number of nearby shallow wells that are used for domestic drinking water purposes (Cumming 1997). The source of the deep upwelling CO2 remains uncertain, but normal faulting associated with the Rio Grande rift likely facilitates upwelling of deep fluids from Paleozoic sedimentary rocks (Cumming 1997). For this study, the focus is documenting and interpreting the measurable effects of CO2 on shallow groundwater chemistry, with particular emphasis on trace elements (F, U, Pb, As, and Fe).
Hydrogeologic setting
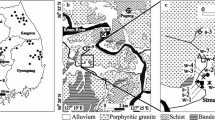
The community of Chimayό, New Mexico lies near the eastern margin of the Española Basin. Formed by extension associated with the Rio Grande rift, the Española Basin is semi-arid, with rainfall ranging from 20 cm/year in the lower elevations to approximately 64 cm/year in the Sangre de Cristo Mountains to the east. Upper Oligocene to upper Miocene basin-fill dominated by sandstone comprises the Tesuque Formation of Santa Fe Group and serves as the aquifer for many communities including Chimayό (Fig. 2). These sediments are quartz and feldspar rich; approximately 50–70% of the felspars are plagioclase (Cavazza 1986). Locally, secondary calcite is abundant and so regional groundwaters are typically in equilibrium with this mineral (Keating and Warren 1999). Rock/water reactions, including chemical weathering of calcite and plagioclase and precipitation of smectite, have been shown to explain trends in major ion chemistry in the regional aquifer (Hereford et al. 2007; Keating and Warren 1999). Cross-sections suggest the aquifer is fairly thin in the vicinity of Chimayό (200–400 m thick; Fig. 3). A wildcat oil well (Castle Wigzell Kelley Federal No. 1), located 3.5 km to the southwest of Chimayo, encountered Pennsylvanian carbonate and siltstone beneath the Tesuque Formation. It is likely that Pennsylvanian strata extend northeast to Chimayό, but reverse faulting from Laramide tectonism may have facilitated local erosional stripping of Pennsylvanian strata under Chimayό; similar erosion from Laramide uplift has been interpreted 10 km to the west (Baldridge et al. 1994). Both the Santa Fe Group and underlying carbonates are dissected by numerous north-south trending faults created by tectonic extension, some of which are cemented with calcite and/or siliceous minerals. Geologic mapping to the south suggests Proterozoic crystalline rocks directly underlies the Pennsylvanian strata (Koning et al. 2002).

Map of Chimayό showing surface geology and well locations (modified from Koning et al. 2002; Koning 2003). Lithosomes A and B refer to sediment associated with an alluvial slope and basin-floor depositional environment, respectively (Cavazza 1986; Smith 2000; Kuhle and Smith 2001; Koning et al. 2005)

Cumming (1997) conducted sampling of wells in the Chimayό community and noticed a strong tendency for shallow wells (depth < 60 m) located near a particular fault (referred to as the Roberts fault in Figs. 2 and 3) to be enriched in dissolved CO2. Cumming (1997) generated a relatively large geochemical dataset (18 wells, most of which were sampled 4 times over a 2-year period). For all but two of the wells, only major ion chemistry is available.
One well in the community geysers approximately twice a day, releasing 99.9% pure CO2. Although the geyser discharge is cyclical, the rate of influx of CO2 into the aquifer from depth may be relatively constant (Lu et al. 2005). Eruption frequency is a function of recharge rate (influx to the bottom of the pipe), and the ratio of length to diameter of the pipe. The fact that only this particular well acts as a geyser may be due to it penetrating a fault-sealed pocket of the aquifer that has locally resulted in high \( {\text{P}}_{{{\text{CO}}_{ 2} }} . \)
At this site, the source of the CO2 recharging the aquifer from below is unclear. Based on limited measurements of carbon isotopes, Cumming (1997) suggested that although the data are inconclusive, the most likely source was mantle CO2. Another possibility, although discounted by the author based on temperature and pressure conditions in the basin, is thermal decarbonation of carbonate rock layers below the Santa Fe Group aquifer A third possibility is dissolution of carbonate rocks by acidic groundwater (a non-CO2 source of acidity). This possibility seems remote because no such waters have been measured in the Española Basin. Without more detailed data collection it is impossible to definitively identify the CO2 source.
There are three mechanisms which could allow the water in the geysering well to rise: (1) CO2 rising as buoyant gas, either as discreet pockets of gas or as diffuse bubbles, (2) upward head gradients in groundwater due to regional driving forces, or (3) thermal buoyancy. If the Roberts fault provides a pathway for CO2 to move upward from depth, it is unclear exactly how the CO2 moves within the fault zone. In this paper, neither the source of the CO2 nor the mechanism for upward movement is addressed. Rather, the focus is on the impact of the CO2 on shallow groundwater chemistry.
To provide context for this study, published groundwater chemistry datasets from 200 wells in the Española Basin, east of the Rio Grande, were combined and plotted on a Durov diagram (Fig. 4) (McQuillan and Montes 1998; USGS 1997; Wust et al. 2005; Street and Finch 2004). These regional groundwaters are generally either Ca-HCO3 or Na-HCO3-type waters. Median values of pH, Cl, and TDS are 8.0, 12.5, and 467 mg/l, respectively. As is evident in Fig. 4, compared to typical regional groundwaters, samples collected by Cumming (1997) in the Chimayo community aquifer tend to be significantly lower in pH and, in some wells, much higher in TDS. Due to the semi-arid environment, thick basin-fill (up to 3–4 km west of the Chimayo area; Cordell 1979), and heterogeneity resulting from faults and dipping strata of various permeabilities, groundwater in the Española Basin tends to move relatively slowly, leading to very long residence times [in some cases, tens of thousands of years (Anderholm 1994; Rogers et al. 1996)], and is frequently enriched in naturally-occurring trace elements including fluoride, uranium, and arsenic (Finch 2005; Gallaher et al. 2004; McQuillan and Montes 1998; McQuillan et al. 2005; Purtymun 1977). Warm springs (<54°C) are present approximately 25 km to the north of Chimayό. These Na-HCO3 type waters, enriched in Li, B, and F, are thought to be upwelling from depth along faults (Vuataz et al. 1984). Cumming (1997) reported that temperatures in Chimayό groundwaters range from 7 to 24°C. Interestingly, although these waters are significantly cooler than the warm springs to the north, they are significantly higher in TDS [over 6,000 mg/l (Cumming 1997) versus 3,600 mg/l] and more enriched in Cl. Br/Cl ratios in the geyser waters are similar to seawater. All waters sampled by Cumming (1997) were oxidizing, with measured dissolved oxygen ranging from 0.4 to 9.3 mg/l. This is typical for the regional aquifer, where due to low availability of organic carbon and thick unsaturated zones (>300 m in some locations) oxygen availability is rarely exceeded by microbial demand. Exceptions to this include local reducing conditions related to domestic septic effluent (personal communication, McQuillan 2003). Data collected by Cumming (1997) show a tendency for groundwater in and near the geysering well to be lower in temperature and dissolved oxygen concentrations than other wells in the vicinity.

Durov diagram, indicating major ion chemistry, pH, and TDS of groundwater in the Espanola Basin, east of the Rio Grande in comparison to groundwater in the Chimayό area (Cumming 1997). Red star (Sanctuario sample) is shown to represent background chemistry
Geochemical sampling
The goal for groundwater sampling was to expand the number of domestic drinking water wells originally sampled by Cumming (1997) and to analyze for major ions, trace elements, and stable isotopes (13C, 18O, 2H). The number of wells available for sampling was limited, however, because many homeowners in the community have recently abandoned their wells and connected to a community water supply. In total, 17 wells were sampled (Fig. 2), four of which were also sampled by Cumming (1997) including the geyser well (#17). The combined dataset represents 31 wells.
Slightly more than half of the samples were collected from hose bibs, the rest were collected at the kitchen tap. When possible, the water was allowed to flow for one minute before sampling. All the samples were untreated and came directly from the well. One sample (#2) was taken from a storage tank where groundwater and surface water are mixed. Sample bottles were filled to the rim and capped within seconds of collection to minimize exposure to air. Samples were tested for pH within one to 2 h of collection using a Thermo Electron Corporation probe. Samples for metal analysis were passed through a 0.05 μm filter. Samples were analyzed for major anions using Ion Chromotography, for cations and trace metals using ICP-MS, for alkalinity using acid titration, and for stable isotopes using a multiflow unit connected to an isotope ratio mass spectrometer.
Sampling results
The chemical data were analyzed using the geochemical modeling code (PHREEQC) (Parkhurst and Appelo 1999) and partial pressure of CO2 (\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)) was calculated for all the sampled wells based on measured pH and alkalinity. All these PHREEQC calculations assume a temperature of 25°C and pe value of 4. For reference, atmospheric \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) = 10−3.5 atm. The results of these calculations and all other chemical analyses are presented in Tables 1, 2, 3. Sixteen of the seventeen samples have calculated log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)) greater than or equal to −2.5; fifteen samples were log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)) > −2.0. This suggests CO2 degassing during sampling did not significantly affect pH measurements (and hence \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) calculations). At the well with the lowest calculated \( {\text{P}}_{{{\text{CO}}_{ 2} }}, \) pH was re-measured and confirmed to be 8.5. This well is used below to represent background conditions, unaffected by CO2 upwelling.
To examine spatial and temporal variation in major ions, this dataset was combined with older data presented by Cumming (1997). Combining these datasets, collected several years apart, does introduce the possibility that temporal trends could be misinterpreted as spatial trends. However, spatial trends in \( {\text{P}}_{{{\text{CO}}_{ 2} }},\) TDS, and [Cl] (Fig. 5) are generally consistent in the two datasets and so temporal variability is presumably smaller than spatial variability. Values of \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) significantly larger than atmospheric (up to 10−2.0 atm) could possibly be caused by biologic activity in the vadose zone during recharge; however, this increase would then typically be offset by mineral weathering reactions in the aquifer. Because organic matter content tends to be low in this setting, we presume waters with \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) greater than 10−2 atm represent groundwater impacted to some degree by influx of CO2 from depth (beneath the aquifer). There are two clusters of wells with very high \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) (Fig. 5a); one near the south end of the minor fault (Roberts fault) and the other where this minor fault projects northwards under Quaternary alluvium to the Chimayό fault. Wells near the Roberts fault (within approximately 500 m of the fault trace) tend to have higher \( {\text{P}}_{{{\text{CO}}_{ 2} }},\) (up to 1 atm) than wells farther from the fault. Not all wells near the fault have high \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) which may be due to complexities in the path of the upwelling \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) and/or mixing and buffering reactions. The wells near the fault trace (within ~500 m) have higher TDS (up to 7,000 mg/l) (Fig. 5b). On the other hand the concentration of chloride is much higher at the southern end of the fault than other locations (Fig. 5c). The increased levels of chloride are important to note as there is no local source for chloride in the shallow aquifer. Since there are no evaporate minerals within the Santa Fe Group, the spatial trends in \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) and [Cl] in Fig. 5 suggest that CO2 is upwelling with deeper brackish waters near the southern end of the fault and is upwelling without deeper brackish waters near the northern end of the fault. Two different relationships between chloride and total carbon (CT = H2CO *3 + HCO3 −) are evident in the data, as shown in Fig. 6. The lowest chloride and CT well samples (lower left corner of Fig. 6) represent dilute background waters, presumably recharged from meteoric sources either in the Sangre de Cristos Mountains to the east or locally along the Santa Cruz River. These have relatively high pH and low [Cl] typical of other waters in the basin. In most of the wells sampled, however, the waters have been impacted to some extent by either CO2 from depth or a CO2-brackish water mixture from depth. Depending on the degree of mixing and mineral precipitation/dissolution reactions in the aquifer, background waters either evolve toward high total carbon/low chloride waters (Path 1, Fig. 6) or high total carbon/high chloride waters (Path 2, Fig. 6). Waters along Path 1 tend to have calcium and bicarbonate as their major cation and anions, respectively. Waters along Path 2, in contrast, are dominated by sodium and chloride. These two cases have very different implication for trace element mobilization and transport, as will be described below.

Variation Chimayό groundwater; a log (\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)), b total dissolved solids, c [Cl]

Variation of chloride and log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)) in Chimayό waters. Solid lines refer to two proposed mixing/geochemistry models, described in text
The stable isotope measurements are shown in Figs. 7 and 8. In contrast to a large number of groundwaters sampled in the region which plot relatively close to the world meteoric line (WML) (Anderholm 1994), Chimayό waters have δ18O/δD ratios slightly shifted to the right (Fig. 7). This can either be interpreted to reflect evaporation or deep circulation (long residence times and/or high temperatures). Stable isotopes measured in cold, meteoric groundwaters in this basin generally show no evidence of evaporation (Anderholm 1994; Blake et al. 1995) and it is difficult to imagine a scenario where Chimayό groundwaters have experienced significant evaporation. A more likely scenario involves mixing of meteoric waters with deeper waters that had very long residence times in host rocks, similar to Path 2 in Fig. 6. This explanation was suggested by Vuataz et al. (1986) for a subset of waters in the Jemez Mountains (to the west) with similar departures from the WML.

Variation in δ18O‰ and δD‰ in Chimayό groundwater. World meteoric line is shown for comparison

Variation in a Cl and b log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)) with respect to δ13C in Chimayό groundwater
As shown in Fig. 8a, waters with higher \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) values tend to be relatively enriched in 13C. We would expect dissolved carbon in meteorically-recharged background waters to be isotopically light (−12 to −15‰) and deeper sources of carbon (either mantle and/or marine-carbonate derived) to be isotopically heavy (−4 to +4‰). Therefore, qualitatively this trend is consistent with the deep water mixing model presented above. In addition to the deep waters being isotopically heavy, they may become further enriched in 13C by degassing CO2 as they upwell. Unless waters are fairly acidic (pH < 5.4), CO2 gas exsolved from groundwater will be isotopically lighter than the remaining dissolved carbon, as observed in Mammoth Lake groundwaters (Evans et al. 2002) and in laboratory experiments (Mook et al. 1974). In the geyser well samples, Cumming (1997) reported a value of δ13C = −7.11 for CO2 gas relative to −0.55‰ in dissolved carbon.
The relationship between chloride and δ13C, shown in Fig. 8a, clearly indicates two populations of waters, as was evident in Fig. 6. The CO2-rich brackish waters near the south end of the Roberts fault are more enriched in 13C and much higher in chloride than all the other waters. Two possible explanations are as follows. Compared to upwelling CO2 in locations other than the south end of the Roberts fault, the upwelling CO2-rich brackish waters either originated at depth at higher pressures, and thus degassed more as they rose, or interacted with isotopically heavier marine carbonate rocks (or both). The possible relationship between higher pressures, more intense degassing, and higher salinity will be explored in future work.
Discussion of transport mechanisms and geochemical modeling
The spatial variability in levels of dissolved CO2 in Chimayό groundwater (Fig. 5a) could be caused by a number of factors: (1) spatial variability in the flux rate of CO2 into the aquifer (regardless of its source) (2) heterogeneity within the aquifer which causes CO2 to build up in some locations, perhaps because of fault-sealing, and to diffuse to the vadose zone quickly in other locations, or (3) spatially-variable CO2-consuming reactions such as calcite or plagioclase dissolution. In addition to spatial variation in CO2, there is large variation in chloride concentrations. This variation is likely due to processes occurring below the aquifer, as CO2 diffuses through older sediments which are heterogeneous. Understanding the role that mineral precipitation/dissolution plays within the aquifer is important to risk assessment studies because mineral alteration will tend to buffer pH changes. This potentially reduces risk to human health since trace element mobility, caused by pH reduction, may be prevented. A negative consequence, however, is that detection of CO2 leakage via monitoring groundwater pH will be more difficult.
To examine the role of carbonate mineral precipitation/dissolution plays in controlling major ion chemistry within the aquifer, geochemical models were constructed using the geochemical modeling code PHREEQC and results were compared to measured trends in water chemistry.
The water chemistry data were separated into the two groups described above (brackish, [Cl] >100 mg/l, and non-brackish, [Cl] <100 mg/l). First, the progressive reaction of CO2 with background water (represented by Well 15, Table 1) was modeled, and results were compared to measured variation in pH, HCO3, \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) in the low [Cl] waters. Possible dissolution/precipitation of calcite, which is ubiquitous as a secondary mineral in these sediments, was also considered. Chemical weathering of plagioclase and other silicates is also known to occur in this basin (Cavazza 1986; Hereford et al. 2007; Keating and Warren 1999), however, dissolution rates are known to be much slower than for carbonate minerals and thus their impact would be a second-order effect. This is supported by calculated saturation indices for groundwaters in Chimayό, which show that these waters are generally near or at equilibrium with respect to calcite and strongly undersaturated with respect to commonly occurring silicate minerals. Two cases were considered:
-
Model A: stepwise reaction of background groundwater with increasing amounts of CO2, while maintaining equilibrium with calcite,
-
Model B: stepwise reaction of background groundwater with increasing amounts of CO2, with no mineral reactions.
Results of these calculations are shown in Fig. 9, in comparison to measured chemical compositions for all waters with low chloride concentrations (<100 mg/l). The data show a tendency for [Ca] to increase and for pH to decrease with increasing \( {\text{P}}_{{{\text{CO}}_{ 2} }}.\) Model B (no calcite dissolution) greatly underpredicts calcium concentrations and overpredicts pH depression. In contrast, Model A matches the data reasonably well, except for at the highest \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) values where predicted [Ca] is too high. Sensitivity studies showed that this discrepancy may be due to neglecting other mineral weathering reactions, such as feldspar dissolution, which may become important at the highest \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) values. Other than these highest \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) calculations, departures of modeled trends from measured trends are not systematic and so are presumably caused by a combination of measurement errors (particularly pH) and natural variability in background water composition, aquifer mineralogy, and mineral dissolution rates. It is noteworthy that a significant percentage of the measured variability in pH, \( {\text{P}}_{{{\text{CO}}_{ 2} }},\) and calcium of these low chloride waters can be described by two simple reactions: influx of \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) and dissolution of calcite.

PHREEQC simulations of background waters reacting with CO2 in equilibrium with calcite compared to measured values of log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)), pH, and Ca for all groundwater samples with [Cl] <100 mg/l
A similar approach was used to determine the role of mineral alteration as upwelling CO2-rich brackish water flows through the shallow aquifer. In this case, the progressive mixing of CO2-rich brackish water (represented by the geyser well) with background water (represented by Well 15), with and without buffering reactions, was modeled. The results of these calculations are shown in Fig. 10, where Model C is mixing and calcite equilibrium and Model D is mixing alone. The measured trends in \( {\text{P}}_{{{\text{CO}}_{ 2} }},\) Ca, pH, and Cl are fairly well reproduced by either of these simple models which yield very similar results. However, influence of mineral dissolution does not appear to be significant. This is because the CO2-rich brackish water is supersaturated with respect to calcite (Table 4) and so as it mixes with background waters it does not significantly enhance calcite dissolution. Small departures from equilibrium with calcite do occur at specific mixing fractions; these produce the small differences between the solid and dashed lines in Fig. 10.

PHREEQC simulations of background waters mixing with CO2 and brackish water (represented by the geyser sample) compared to water samples with [Cl] > 100 mg/l. a measured and simulated pH and [Cl]; b measured and simulated [Ca]
Finally, a possible explanation for the major ion chemistry measured in the geyser well was tested using PHREEQC. The conceptual model proposed by Cumming (1997) of a connate brine rising with CO2 through the carbonate strata underlying the aquifer was considered. Using a pure Na–Cl brine as a starting composition, titration with CO2 in equilibrium with carbonate (calcite and dolomite) was simulated until the \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) measured at the geyser well was achieved. Waters were allowed to be slightly supersaturated with respect to carbonate minerals, as reflected in calculated saturation indices in geyser well samples (see Table 4). This apparent supersaturation may be due to sampling biases caused by degassing, or may reflect in situ disequilibrium due to rapid temperature changes as CO2-rich water rises in the well and degasses. By adjusting brine alkalinity and target saturation indices, good agreement between simulated and measured geyser well chemistry was achieved, as shown in Fig. 11. The range of simulated values (bars) represents variation that would be expected due to \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) variation alone. Model-assumed values for the endmember brine were [HCO3−] = 1613 mg/l, SI (calcite) = 0.8, SI (dolomite) = 0.6. Departures from simulated and measured values may have been caused by variations other than \( {\text{P}}_{{{\text{CO}}_{ 2} }},\) such as variations in brine chemistry with time or by reactions unaccounted for in this simple model.

Measured concentrations of major ions in the geyser well (open symbols); range of simulated values (bars)
Although there are probably other geochemical models that would be equally consistent with the measured water chemistry data, the very simple models tested here are generally consistent with observed trends at this site and known aquifer mineralogy found in this area. We conclude that when CO2 diffuses through the aquifer, it reaches higher levels in some locations than in others due to geologic heterogeneity, hydrodynamic factors, and/or spatial variations in CO2 flux. Carbonate dissolution is sufficiently rapid relative to the CO2 flux rate so as to prevent pH from lowering below 6.2 in the vast majority of samples (note: during one sampling round reported by Cumming (1997) somewhat lower values (≤5.8) were reported in 3 wells). In contrast, when CO2 upwells along with brackish water, the reactivity of the solution is low and mineral weathering is inconsequential. Finally, a conceptual model of CO2-rich connate brine flowing through and dissolving carbonate rocks below the aquifer, as proposed by Cumming (1997), is quantitatively consistent with measured values of major ion concentrations at the geyser well.
Impact of CO2 on trace element concentrations
Evidence from measurements and modeling, described above, suggest that there are two distinct mechanisms controlling major ion and stable isotope chemistry at the site: (1) influx of CO2 and subsequent in situ carbonate mineral dissolution and (2) influx of CO2-rich brackish water with relatively little carbonate alteration. The two groups of waters can be easily distinguished on the basis of chloride content and carbon isotopes. Recognizing the difference between these two mechanisms is critical for interpretation of trace element concentration data, since mechanism (1) has the potential for mobilizing trace metals within the aquifer. This is due to increased levels of CO2 and depressed pH. Mechanism (2) has the additional potential for transporting trace elements into the aquifer. By examination of trace element concentrations in the geyser well (Table 2), it is clear that mechanism (2) has the potential to transport trace elements into the aquifer from below. These elevated trace elements are either associated with the connate brine or with carbonate dissolution in layers below the aquifer.
Of the elements listed in Table 2, the focus of more detailed analysis is trace elements possessing known health effects and which were measured at significant concentrations in one or more samples: Fe, U, Pb, As, and F. Variations of these trace element concentrations with \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) and chloride are shown in Fig. 12. Samples belonging to the two groups (defined based on chloride and carbon isotopes) are indicated by symbols: mechanism (1) (black symbols) and mechanism (2) (red symbols). Although our sample size is too small to allow statistically robust comparisons, several notable trends are evident. Figure 12a suggests an apparent tendency for high \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) water to be enriched in Fe, Pb, As, and U, which might at first glance imply that CO2 is mobilizing trace metals. However, three of these elements (Pb, As, and U) are linearly correlated with [Cl] in the high-chloride waters (Fig. 12b); suggesting instead (1) the source of these elements is the deep brackish waters and (2) they behave conservatively during mixing with shallow fresh water (Path 2, Fig. 6). This evidence, combined with the lack of a relationship between \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) and [U] and [Pb] in low [Cl] waters, suggests that CO2 is not driving in situ reactions in the shallow aquifer and is not mobilizing these elements. PHREEQC calculations were used to examine possible reasons why these elements are apparently non-reactive. All the samples are strongly undersaturated with respect to common U and Pb-bearing minerals (Table 4). In total, this indicates that mineral dissolution, if occurring at the site, is very slow relative to the flux of groundwater and CO2. In particular the geyser water, which has the highest concentration of U, is significantly undersaturated with respect to uraninite. To test the possibility that this water might have been in equilibrium with uraninite at warmer temperatures at depth, PHREEQC calculations were repeated at a much higher temperatures and presumed reducing conditions. Simple geothermometers based on Na, K, Ca, and Si (Fournier and Truesdell 1973; Fournier 1979; Fournier and Potter 1982) would indicate original temperatures in the range of 100–140°C for the geyser water. PHREEQC results show that for values of pe ≤ 0, the geyser water would have been either at or above saturation with respect to uraninite. Among low [Cl] waters, there is no relationship between measured [HCO3 −] and [U] or [Pb] and so carbonate complexation is not significantly enhancing dissolution or transport. There is no relationship between [U], [Pb] and the presence/absence of [NO3] or [Fe]. Therefore, redox state does not appear to control variability.

a Trace element concentrations, in mg/l, in relation to calculated log(\( {\text{P}}_{{{\text{CO}}_{ 2} }} \)). Dotted lines indicate EPA primary drinking water standards (not applicable to Pb and Fe). b Trace element concentrations, in mg/l, in relation to [Cl] (mg/l). Dotted lines indicate EPA primary drinking water standards (not applicable to Pb and Fe)
Similar arguments apply to observed trends in [As]. Like uranium, arsenic appears to behave conservatively during mixing of high-[As] brackish water and background water. Interestingly, one low-[Cl] sample has significant levels of arsenic. PHREEQC results indicate that arsenic exists predominately as a neutral species (H3AsO3), and is very far from equilibrium for any arsenic-containing minerals. Therefore, precipitation/dissolution reactions must be very slow or non-existent and sorption/desportion is unlikely due to the neutral charge of the aqueous complex. Interestingly, waters are supersaturated with respect to Ba3(AsO4)2, and at or near equilibrium with barite (BaSO4). Geochemical interactions between barium, arsenic, and associated minerals may be important at this site.
Fluoride exists almost exclusively as F- in these waters, according to PHREEQC calculations. Previous studies have suggested that F participates in ion exchange; clays are known to adsorb F at low or moderate pH’s (Saxena and Ahmed 2003; Finch 2005). The very high F concentration in the highest pH sample (# 14) lends credence to this possibility. If pH and adsorption/desorption are controlling [F], then CO2 flux could be affecting fluoride geochemistry. Additionally, several samples are near equilibrium with fluorite, suggesting relatively fast precipitation/dissolution reactions. Apparently fluoride is relatively reactive in this environment, but there is clearly no tendency for enhanced \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) to increase [F].
Of all these elements, iron appears to be the most reactive. Like fluoride, however, there is no evidence that increased \( {\text{P}}_{{{\text{CO}}_{ 2} }} \) leads to higher levels of [Fe]. As shown in Fig. 12, [Fe] concentrations are negligible in low-[Cl] waters, regardless of \( {\text{P}}_{{{\text{CO}}_{ 2} }}.\) Many samples are at or near equilibrium with respect to iron hydroxides and so rapid precipitation and/or dissolution of amorphous iron minerals may be occurring at this site. Iron is also likely to participate in sorption/desorption reactions. The charge of the dominant aqueous iron species varies with pH at this site; Fe(OH)+ dominates in lower pH waters and Fe(OH)3 dominates in neutral or basic pH waters. Therefore, iron may preferentially sorb to mineral surfaces in waters most impacted by CO2. Redox conditions, probably unrelated to CO2, may the important as well. This is supported by a negative correlation between dissolved iron and nitrate which are, in many samples, mutually exclusive. The very high concentrations in Well 16 may be due to a combination of local redox conditions and well pipe corrosion.
Conclusions and implications for monitoring at a CO2 sequestration site
From this relatively small sample size at a natural analog site, it is clear that CO2 is upwelling via at least two pathways; one without significant entrainment of brackish water and one with brackish water. The latter pathway provides a source of arsenic, uranium, and lead into the aquifer that, while significant, should not be confused with in situ trace element mobilization. There is no evidence in this dataset that CO2 is mobilizing arsenic, uranium, or lead from minerals within the shallow aquifer. Our geochemical modeling suggests dissolution of calcite buffers against significant pH depression and may, therefore, inhibit trace metal mobilization. This is in agreement with reactive-transport modeling studies (Wang and Jaffe 2004) which predict low or zero enhanced trace metal mobility in buffered, high alkalinity aquifers.
There is some indication, however, that CO2 transport is affecting fluoride geochemistry within the aquifer. In this environment, the CO2-induced pH depression may be causing fluoride to be adsorbed into aquifer sediments via ion exchange. In aquifers such as this one where naturally-occurring fluoride is a significant hazard locally, CO2 has a beneficial affect on groundwater quality. There are no clear trends with iron concentrations, which generally tend to be very low in this oxidizing environment.
There are several implications for CO2 leakage impacts. First, as is evident at the Chimayό site, brine that might either leak directly from the CO2 reservoir or be entrained into the CO2 plume as it passes through rocks above the reservoir could have a much greater impact on shallow groundwater quality than the CO2 itself or by mineral reactions in the aquifer driven by elevated CO2. As was suggested by Kharaka et al. (2006), consideration of brine co-leakage and/or generation of brines through CO2-induced dissolution should be an important element of risk assessment. Second, there are a number of factors that could mitigate the impact of CO2 leakage on shallow groundwater quality and thus make the CO2 leak difficult or impossible to detect. These include (1) simple mixing and dilution of CO2-impacted groundwater with ambient groundwater, (2) pH buffering reactions such as calcite dissolution and/or silicate mineral weathering, (3) limited trace metal availability in aquifer minerals, and (4) trace metal scavenging by secondary mineral precipitation. Evaluating the potential importance of the first mechanism, mixing, will require knowledge of the possible mechanisms of leakage (diffuse or focused), the relative magnitude of the leakage flux and ambient groundwater fluxes, and some understanding of local groundwater flow dynamics. Simple calculations and relatively inexpensive site characterization could be very useful for this purpose. Evaluating the potential importance of the buffering reactions due to mineral weathering should also be relatively easy to assess with limited site information. The last two geochemical factors will be very difficult to evaluate without very detailed, site-specific geochemical characterization and even then, the limited predictive capability of available geochemical models may not be adequate for risk assessment purposes. Fortunately, if the first two factors can be shown to lessen the risk, as appears to be the case in the Chimayό aquifer system, there may be no need to address the more complex and inherently site-specific geochemistry. Conditions favorable for pH depression include a lack of buffering minerals, hydrologic conditions that minimize mixing and dilution of high CO2 water (spatially focused, high CO2 flux), and aquifer heterogeneities that can trap CO2-enriched water locally and inhibit rapid degassing to the vadose zone. If these conditions exist, very detailed site-specific characterization and monitoring will be critical for any meaningful risk assessment.
References
Aiuppa A, Federico C, Allard P, Gurrieri S, Valenza M (2005) Trace metal modeling of groundwater–gas–rock interactions in a volcanic aquifer: Mount Vesuvius, Southern Italy. Chem Geol 216:289–311
Anderholm SK (1994) Ground-water recharge near Santa Fe, north-central New Mexico, USGS Water Resources Investigations Report 94-4078, 68 pp
Baldridge WS, Ferguson JF, Braile LW, Wang B, Eckhardt K, Evans D, Schultz C, Gilpin B, Jiracek GR, Biehler S (1994) The western margin of the Rio Grande Rift in northern New Mexico: An aborted boundary? GSA Bull 105:1538–1551
Benson SM (2002) Lessons learned from natural and industrial analogues for storage of carbon dioxide in deep geological formations. Lawrence Berkeley Laboratory Report, 227 pp
Birkholzer JT (2008) Prediction of potential groundwater contamination in response to CO2 leakage from deep geological storage, 7th annual carbon sequestration conference: Pittsburgh, PA
Birkholzer J, Zhou Q, Rutqvist J, Jordan P, Zhang K, Tsang C-F (2007) Research project on CO2 geological storage and groundwater resources: Large-scale hydrological evaluation and modeling of the impact on groundwater systems., Annual Report Cot 1, 2006-Sept 30-2007. National Energy Technology Lab
Blake WD, Goff F, Adams AI, Counce D (1995) Environmental Geochemistry for surface and subsurface waters in the Pajarito Plateau and outlying areas, New Mexico, Los Alamos National Laboratory Report, 43 pp
Bose P, Sharma A (2002) Role of iron in controlling speciation and mobilization of arsenic in subsurface environment. Water Res 36:4916–4926
Bruno J, Stumm W, Wesin P, Bandberg F (1992) On the influence of carbonate in mineral dissolution: I. the thermodyamics and kinetics of hematite dissolution in bicarbonate solutions at T = 25°C. Geochim Cosmochim Acta 56:1139–1147
Cavazza W (1986) Miocene sediment dispersal in the Central Espanola Basin, Rio Grande Rift, New Mexico, USA. Sed Geol 5:119–135
Chen Z (2005) In situ feldspar dissolution rates in an aquifer. Geochimca et Cosmochimica Acta 69:1435–1453
Craw D, Falconer D, Youngson JH (2003) Environmental arsenopyrite stability and dissolution: theory, experiment, and field observations. Chem Geol 199:71–82
Cumming KA (1997) Hydrogeochemistry of groundwater in Chimayo, New Mexico M.S., Northern Arizona University, Flagstaff, AZ, 117 pp
Evans WC, Sorey ML, Cook AC, Kennedy BM, Shuster DL, Colvard EM, White LD, Huebner MA (2002) Tracing and quantifying magmatic carbon discharge in cold groundwaters: Lessons learned from Mammoth Mountain, USA. J Volcanol Geoth Res 114:291–312
Federico C, Aiuppa A, Favara R, Gurrieri S, Valenza M (2004) Geochemical monitoring of groundwaters (1998–2001) at Vesuvius volcano (Italy). J Volcanol Geoth Res 133:81–104
Finch ST (2005) Occurrence of elevated arsenic and fluoride concentrations in the Española Basin. In: McKinney KC (ed) 4th Annual Española basin workshop. U.S.Geological Survey, Santa Fe, p 33
Fournier RO (1979) Revised equation for the Na/K geothermometer. In: Transactions−Geothermal Resources Council. Geothermal Resources Council, Davis, pp 221–224
Fournier RO, Potter RWII (1982) Revised and expanded silica (quartz) geothermometer. Bull Geotherm Resour Counc (Davis, Calif) 11(10):3–12
Fournier RO, Truesdell AH (1973) Empirical Na–K–Ca geothermometer for natural waters. Geochim Cosmochim Acta 37(5):1255–1257
Gallaher BM, Efurd DW, Steiner RE (2004) Uranium in waters near Los Alamos National Laboratory: concentrations, trends, and isotopic composition through 1999. Los Alamos National Laboratory, Los Alamos, 70 pp
Glennon JAaPRM (2005) The operation and geography of carbon-dioxide-driven, cold-water geysers. GOSA Trans 9:184–192
Goff F, Shevenell L, Gardner JN, Vuataz FD, Grigsby CO (1988) The hydrothermal outflow plume of Valles Caldera, New Mexico, and a comparison with other outflow plumes. J Geophys Res 93:6041–6058
Golubev SV, Pokrovsky OS, Schott J (2005) Experimental determination of the effect of dissolved CO2 on the dissolution kinetics of Mg and Ca silicates at 25°C. Chem Geol 217:227–238
Hereford AG, Keating E, Guthrie GD, Zhu C (2007) Reactions and reaction rates in the regional aquifer beneath the Pajarito Plateau, north-central New Mexico, USA. Env Geol 52:965–977
Keating E, Warren R (1999) Geochemistry of the regional aquifer, Los Alamos National Laboratory Report, 35 pp
Kharaka YK, Cole DR, Hovorka S, Gunter WD, Knauss KG, Freifeld BM (2006) Gas–water–rock interactions in Frio Formation following CO2 injection: implications for the storage of greenhouse gases in sedimentary basins. Geology 34:577–580
Koning DJ (2003) revised Dec-2005, Geologic map of the Chimayo 7.5-minute quadrangle, Rio Arriba and Santa Fe Counties, New Mexico: New Mexico Bureau of Geology and Mineral Resources, Open-file Geologic Map OF-GM-71, scale 1:24,000
Koning DJ, Skotnicki S, Nyman M, Horning R, Eppes M, Rogers S (2002) Geology of the Cundiyo 7.5-minute quadrangle, Santa Fe County, New Mexico: New Mexico Bureau of Geology and Mineral Resources, Open-file Geologic Map OF-GM-56, scale 1:24,000
Koning DJ, Connell SD, Morgan GS, Peters L, McIntosh WC (2005) Stratigraphy and depositional trends in the Santa Fe Group near Española, north-central New Mexico: tectonic and climatic implications: New Mexico Geological Society, 56th Field Conference Guidebook, Geology of the Chama Basin, pp 237–257
Kuhle AJ, Smith GA (2001) Alluvial-slope deposition of the skull ridge member of the Tesuque formation, Española Basin. NM NM Geol 23:30–37
Langmuir D (1997) Aqueous Environmental Chemistry: Upper Addle River, New Jersey. Prentice-Hall, Englewood Cliffs, 600 pp
Liewicki JL, Birkholzer JT, Tsang C-F (2007) Natural and industrial analogues for leakage of CO2 from storage reservoirs: identification of features, events, and processes and lessons learned. Env Geol 52:457–467
Lu X, Watson A, Gorin AV, Deans J (2005) Measurements in a low temperature CO2-driven geysering well, viewed in relation to natural geysers. Geothermics 34:389–410
McQuillan D, Montes R (1998) Ground-water geochemistry, Pojoaque Pueblo, New Mexico: Santa Fe, NM, New Mexico Environment Department, 34 pp
Mook WG, Bommerson JC, Staverman WH (1974) Carbon isotope fractionation between dissolved bicarbonate and gaseous carbon dioxide. Earth Planet Sci Lett 22:169–176
Newell D, Crossey LJ, Karlstrom KE, Fischer TP (2005) Continental-scale links between the mantle and groundwater systems of the western United States: Evidence from travertine springs and regional He isotope data. GSA Today 15:4–10
Purtymun WD (1977) Hydrologic characteristics of the Los Alamos Well field, with reference to the occurrence of arsenic in well LA-6, Los Alamos National Laboratory Report, 63 pp
Rogers DB, Stoker AK, McLin SG, Gallaher BM (1996) Recharge to the Pajarito Plateau regional aquifer system, NM Geol Soc Guidebook, 47th Field conference, Jemez Mountins Region, pp 407–412
Rogie JD, Kerrick DM, Sorey ML, Chiodini G, Galloway DL (2001) Dynamics of carbon dioxide emission at Mammoth Mountain, California. Earth Planet Sci Lett 188:535–541
Saxena VK, Ahmed S (2003) Inferring the chemical parameters for the dissolution of fluoride in groundwater. Env Geol 43:731–736
Smith GA (2000) Recognition and significance of stream-flow dominated piedmont facies in extensional basins. Basin Res 12:399–411
Smyth RC, Holtz MH, Guillot SN (2006) Assessing impacts to groundwater from CO2-flooding of SACROC and Claytonville oil fields in West Texas. presented at the 2006 UIC Conference of the Groundwater Protection Council, Austin, Texas, 24 January 2006
Stephens JC, Hering JG (2004) Factors affecting the dissolution kinetics of volcanic ash soils: dependencies on pH, CO2, and oxalate. Appl Geochem 19:1217–1232
Street JB, Finch S (2004) Well report Buckman wells no. 10 through 13. John Shomaker and Associates, Albuquerque, 59 pp
U.S. Geological Survey (1997) WATSTORE database, vol 1997 retrieval
Vuataz FD, Stix J, Goff F, Pearson CF (1984) Low-temperature geothermal potential of the Ojo Caliente warm springs area, northern New Mexico, Los Alamos National Laboratory Report, 56 pp
Vuataz FD, Goff F, Fouillac C, Calvez JY (1986) Isotope geochemistry of thermal and nonthermal waters in the Valles Caldera, Jemez Mountains, northern New Mexico. J Geophysics Res 91:1835–1853
Wang S, Jaffe PR (2004) Dissolution of a mineral phase in potable aquifers due to CO2 releases from deep formations; effect of dissolution kinetics. Energy Convers Manage 45:2833–2848
Acknowledgments
This work was supported by the US DOE through the Zero Emission Research Technology (ZERT) project. We thank Jim Roberts for access to his wells and to all the other residents of Chimayό for allowing us to sample their water. We thank Toti Larson, Emily Kluk, Mike Rearick, and George Perkins for their laboratory analyses and discussions on data interpretation. We also gained invaluable insight from discussions with Bill Carey and Dennis Newell. Andi Kron assisted with illustrations. We appreciate thoughtful discussion with June Fabryka-Martin and the manuscript was greatly improved by the comments from one anonymous reviewer.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Keating, E.H., Fessenden, J., Kanjorski, N. et al. The impact of CO2 on shallow groundwater chemistry: observations at a natural analog site and implications for carbon sequestration. Environ Earth Sci 60, 521–536 (2010). https://doi.org/10.1007/s12665-009-0192-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-009-0192-4