
Abstract
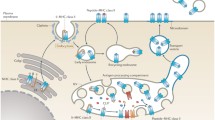
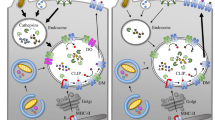
T cells bearing αβ receptors recognize antigenic peptides bound to class I and class II glycoproteins encoded in the major histocompatibility complex (MHC). Cytotoxic and helper T cells respond respectively to peptide antigens derived from endogenous sources presented by MHC class I, and exogenous sources presented by MHC II, on antigen presenting cells. Differences in the MHC class I and class II structures and their maturation pathways have evolved to optimize antigen presentation to their respective T cells. A main focus of our laboratory is on efforts to understand molecular events in processing of antigen for presentation by MHC class II. The different stages of MHC class II—interactions with molecular chaperons involved in folding and traffic from the ER through the antigen-loading compartments, peptide exchange, and transport to the cell surface have been investigated. Through intense research on biophysical and biochemical properties of MHC class II molecules, we have learned that the conformational heterogeneity of MHC class II induced upon binding to different peptides is a key regulator in antigen presentation and epitope selection, and a determinant of the ability of MHC class II to participate in peptide association or dissociation and interaction with the peptide editor HLA-DM.

Similar content being viewed by others

References
Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol. 1993;11:403–50.
Sadegh-Nasseri S, Germain RN. How MHC class II molecules work: peptide-dependent completion of protein folding. Immunol Today. 1992;13(2):43–6.
Sadegh-Nasseri S, Germain RN. A role for peptide in determining MHC class II structure. Nature. 1991;353(6340):167–70.
Sadegh-Nasseri S, McConnell HM. A kinetic intermediate in the reaction of an antigenic peptide and I-Ek. Nature. 1989;337(6204):274–6.
Sadegh-Nasseri S, Stern LJ, Wiley DC, Germain RN. MHC class II function preserved by low-affinity peptide interactions preceding stable binding. Nature. 1994;370(6491):647–50.
Sato AK, Zarutskie JA, Rushe MM, Lomakin A, Natarajan SK, Sadegh-Nasseri S, et al. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J Biol Chem. 2000;275(3):2165–73.
Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry. 2005;44(42):13625–37.
Dornmair K, Rothenhausler B, McConnell HM. Structural intermediates in the reactions of antigenic peptides with MHC molecules. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):409–16.
Witt SN, McConnell HM. Formation and dissociation of short-lived class II MHC-peptide complexes. Biochemistry. 1994;33(7):1861–8.
Schmitt L, Boniface JJ, Davis MM, McConnell HM. Conformational isomers of a class II MHC-peptide complex in solution. J Mol Biol. 1999;286(1):207–18.
Natarajan SK, Assadi M, Sadegh-Nasseri S. Stable peptide binding to MHC class II molecule is rapid and is determined by a receptive conformation shaped by prior association with low affinity peptides. J Immunol. 1999;162(7):4030–6.
Rabinowitz JD, Vrljic M, Kasson PM, Liang MN, Busch R, Boniface JJ, et al. Formation of a highly peptide-receptive state of class II MHC. Immunity. 1998;9(5):699–709.
Zarutskie JA, Busch R, Zavala-Ruiz Z, Rushe M, Mellins ED, Stern LJ. The kinetic basis of peptide exchange catalysis by HLA-dm. Proc Natl Acad Sci USA. 2001;98(22):12450–5.
Watts TH, Brian AA, Kappler JW, Marrack P, McConnell HM. Antigen presentation by supported planar membranes containing affinity-purified I-Ad. Proc Natl Acad Sci USA. 1984;81(23):7564–8.
Buus S, Sette A, Colon SM, Jenis DM, Grey HM. Isolation and characterization of antigen-IA complexes involved in T cell recognition. Cell. 1986;47(6):1071–7.
Dornmair K, McConnell HM. Refolding and reassembly of separate alpha and beta chains of class II molecules of the major histocompatibility complex leads to increased peptide-binding capacity. Proc Natl Acad Sci USA. 1990;87(11):4134–8.
Germain RN, Hendrix LR. MHC class II structure, occupancy and surface expression determined by post-endoplasmic reticulum antigen binding. Nature. 1991;353(6340):134–9.
Davidson HW, Reid PA, Lanzavecchia A, Watts C. Processed antigen binds to newly synthesized MHC class II molecules in antigen-specific b lymphocytes. Cell. 1991;67(1):105–16.
Sadegh-Nasseri S. Peptide, invariant chain, or molecular aggregation preserves class II from functional inactivation. In: Humphreys RE, Pierce SK, editors. Antigen processing and presentation, vol. 1. San Diego: Academic Press; 1994. p. 170–87.
Park SJ, Sadegh-Nasseri S, Wiley DC. Invariant chain made in Escherichia coli has an exposed n-terminal segment that blocks antigen binding to HLA-DR1 and a trimeric c-terminal segment that binds empty HLA-DR1. Proc Natl Acad Sci USA. 1995;92(24):11289–93.
Jardetzky TS, Gorga JC, Busch R, Rothbard J, Strominger JL, Wiley DC. Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J. 1990;9(6):1797–803.
Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368(6468):215–21.
Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure. 1997;5(10):1385–96.
Hammer J, Valsasnini P, Tolba K, Bolin D, Higelin J, Takacs B, et al. Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell. 1993;74(1):197–203.
Wu S, Gorski J, Eckels DD, Newton-Nash DK. T cell recognition of MHC class II-associated peptides is independent of peptide affinity for MHC and sodium dodecyl sulfate stability of the peptide/MHC complex. Effects of conservative amino acid substitutions at anchor position 1 of influenza matrix protein19–31. J Immunol. 1996;156(10):3815–20.
Verreck FA, Vermeulen C, Poel AV, Jorritsma P, Amons R, Coligan JE, et al. The generation of SDS-stable HLA DR dimers is independent of efficient peptide binding. Int Immunol. 1996;8(3):397–404.
Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DA, et al. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature. 1992;358(6389):764–8.
Natarajan SK, Stern LJ, Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J Immunol. 1999;162(6):3463–70.
Stern LJ, Wiley DC. The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell. 1992;68(3):465–77.
Germain RN, Rinker AG Jr. Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature. 1993;363(6431):725–8.
Romagnoli P, Germain RN. The clip region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J Exp Med. 1994;180(3):1107–13.
Chou CL, Sadegh-Nasseri S. HLA-dm recognizes the flexible conformation of major histocompatibility complex class II. J Exp Med. 2000;192(12):1697–706.
Pu Z, Lovitch SB, Bikoff EK, Unanue ER. T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity. 2004;20(4):467–76.
Marin-Esteban V, Falk K, Rotzschke O. “Chemical analogues” Of HLA-DM can induce a peptide-receptive state in HLA-DR molecules. J Biol Chem. 2004;279(49):50684–90.
Pashine A, Busch R, Belmares MP, Munning JN, Doebele RC, Buckingham M, et al. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity. 2003;19(2):183–92.
Belmares MP, Busch R, Mellins ED, McConnell HM. Formation of two peptide/MHC II isomers is catalyzed differentially by HLA-DM. Biochemistry. 2003;42(3):838–47.
Stratikos E, Mosyak L, Zaller DM, Wiley DC. Identification of the lateral interaction surfaces of human histocompatibility leukocyte antigen (HLA)-DM with HLA-DR1 by formation of tethered complexes that present enhanced HLA-dm catalysis. J Exp Med. 2002;196(2):173–83.
Doebele RC, Busch R, Scott HM, Pashine A, Mellins ED. Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity. 2000;13(4):517–27.
Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, et al. HLA-DM targets the hydrogen bond between the histidine at position beta81 and peptide to dissociate HLA-DR-peptide complexes. Nat Immunol. 2007;8(1):92–100.
Fremont DH, HenDRickson WA, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272(5264):1001–4.
Fremont DH, Monnaie D, Nelson CA, HenDRickson WA, Unanue ER. Crystal structure of I-Ak in complex with a dominant epitope of lysozyme. Immunity. 1998;8(3):305–17.
Wilson N, Fremont D, Marrack P, Kappler J. Mutations changing the kinetics of class II MHC peptide exchange. Immunity. 2001;14(5):513–22.
McFarland BJ, Beeson C, Sant AJ. Cutting edge: a single, essential hydrogen bond controls the stability of peptide-MHC class II complexes. J Immunol. 1999;163(7):3567–71.
Saito K, Oda M, Sarai A, Azuma T, Kozono H. Contribution of a single hydrogen bond between betahis81 of MHC class II I-E(k) and the bound peptide to the PH-dependent thermal stability. Microbiol Immunol. 2004;48(1):53–7.
Sadegh-Nasseri S, Chen M, Narayan K, Bouvier M. The convergent roles of tapasin and HLA-DM in antigen presentation. Trends Immunol. 2008;29(3):141–7.
Narayan K, Su KW, Chou CL, Khoruzhenko S, Sadegh-Nasseri S. HLA-DM mediates peptide exchange by interacting transiently and repeatedly with HLA-DR1. Mol Immunol. 2009;46(15):3157–62.
Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. 2007;26(6):1681–90.
Acknowledgments
SS-N wishes to specially thank Dr. Harden McConnell for his remarkable mentorship and for rightfully pointing out the importance of kinetic experiments. Funding for the research described here was from Cancer Research Institute, NIAID and NIGM. This work was supported by R01GM53549 and R01AI063764 grants to SS-N.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sadegh-Nasseri, S., Natarajan, S., Chou, CL. et al. Conformational heterogeneity of MHC class II induced upon binding to different peptides is a key regulator in antigen presentation and epitope selection. Immunol Res 47, 56–64 (2010). https://doi.org/10.1007/s12026-009-8138-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-009-8138-1