
Opinion statement
Central nervous system (CNS)-hemophagocytic lymphohistiocytosis (HLH) is not a disease in itself, but it is part of a systemic immune response. The vast majority of patients with CNS-HLH also have systemic HLH and a large number of patients with primary and secondary HLH have CNS involvement. Reactivations within the CNS are frequent during the course of HLH treatment and may occur concomitant with or independent of systemic relapses. It is also important to consider primary HLH as an underlying cause of “unknown CNS inflammation” as these patients may present with only CNS disease. To initiate proper treatment, a correct diagnosis must be made. A careful review of the patient’s history and a thorough neurological examination are essential. In addition to the blood tests required to make a diagnosis of HLH, a lumbar puncture with cerebrospinal fluid (CSF) analysis and magnetic resonance imaging (MRI) should always be done in all cases regardless of the presence or absence of neurological signs or symptom. Treatment options for CNS-HLH include, but are not limited to, those commonly used in systemic HLH, including corticosteroids, etoposide, cyclosporine A, alemtuzumab, and ATG. In addition, intrathecal treatment with methotrexate and corticosteroids has become a standard care and is likely to be beneficial. Therapy must be initiated without inappropriate delay to prevent late effects in HLH. An interesting novel approach is an anti-IFN-gamma antibody (NI-0501), which is currently being tested. Hematopoietic stem cell transplantation (HSCT) also represents an important CNS-HLH treatment; patients with primary HLH may benefit from immediate HSCT even if there is active disease at time of transplantation, though care should be taken to monitor CNS inflammation through HSCT and treat if needed. Since CNS-HLH is a condition leading to the most severe late effects of HLH, early expert consultation is recommended.
Similar content being viewed by others

Introduction
Hemophagocytic lymphohistiocytosis (HLH) is not a single disease but rather a clinical syndrome of life-threatening hyperinflammation. It may result from genetic defects (primary HLH [1•]) or be acquired with infectious, neoplastic, autoinflammatory, autoimmune, and immunodeficiency etiologies (secondary HLH) [1•]. Primary HLH is rare, with an estimated yearly incidence in Sweden of 0.12–0.15 per 100,000 children [2]. The incidence of primary HLH in adults or that of secondary HLH is not studied.
The term “central nervous system (CNS) disease” is frequently used in the HLH-related literature; but to date, there is no consensus regarding its definition. However, most HLH experts agree that an abnormal CSF and/or MRI of the brain, with or without distinct neurological signs or symptoms, define CNS-HLH. Although differences of opinion still exist on how to define CNS disease, there is agreement that the term refers to infiltration of activated lymphocytes and macrophages into the meninges and brain [3]. CNS disease has been divided into three neuropathological stages: stage I with leptomeningeal inflammation, stage II with perivascular infiltration, and stage III with massive tissue infiltration, blood vessel destruction, and tissue necrosis. This infiltration can induce devastating brain lesions in affected patients and is an important cause of mortality and morbidity in HLH [4••, 5].
In both primary and secondary HLH, CNS involvement is a frequent finding at disease onset [6,6,7,9]. In addition, disease reactivation, during or after therapy, occurs frequently in the CNS. Overall, CNS disease has been reported in 30–73% of all HLH patients, either at presentation or during the course of the disease [7, 10, 11••]. As the disease may be difficult to diagnose, a high index of suspicion is required when evaluating patients with systemic HLH. The clinical picture of HLH is similar in primary and secondary cases and is characterized by systemic inflammation, markedly elevated cytokine levels and immune-mediated organ damage [12, 13••, 14]. HLH is currently diagnosed by either (1) a proven genetic mutation or (2) fulfilling five out of eight clinical criteria (fever, splenomegaly, cytopenias of at least two cell lines, hypertriglyceridemia and/or hypofibrinogenemia, hyperferritinemia, abnormally low NK-cell activity, high levels of soluble IL-2 receptor, and pathologic evidence of hemophagocytosis in tissues) [15]. By contrast, the clinical presentation of CNS disease in HLH is highly variable [8, 16]. Occurrence of neurological symptoms is not included as a diagnostic criterion of HLH, but it is important to consider HLH in a child with unexplained neurologic manifestations, especially one with fever, pancytopenia, and hepatosplenomegaly.
As survival in patients with HLH has improved markedly [7, 17], it has become increasingly important to thoroughly evaluate long-term sequelae, the most important of which are neurological. Unfortunately, significant motor and cognitive deficits may occur following HLH [16, 18••]. Early recognition and prompt treatment of CNS disease may prevent irreversible CNS injury and are therefore of greatest importance in order to improve the long-term outcome [5, 16]. Neurological outcomes after treatment are, however, unknown as therapy trials have tended to merely focus on survival.
To recommend the best possible treatment for CNS involvement, we need to understand the pathophysiology of CNS-HLH. It is likely similar to that of systemic HLH, i.e., massive hyper-inflammation leading to destruction of brain tissue. Therefore, it is important to constantly reduce inflammatory HLH activity to prevent CNS injury. In addition, insufficient cytotoxicity in primary HLH may result in reduced elimination of virus infected cells; hence, antiviral therapy should be given when possible. The HLH therapy is based on specific immunotherapy and/or chemotherapy regimens followed by hematopoietic stem cell transplantation (HSCT) in primary HLH [7, 15, 17].
Here, we review the available literature on treatment of CNS involvement in HLH, including mechanisms of action and clinical efficacy. We also present suggestions regarding treatment of CNS-HLH.
Definition of CNS Disease in HLH
The definition of CNS disease in HLH has not been standardized. Importantly, however, retrospective studies suggest that the presence of CNS disease carries key prognostic significance [19], including higher risk of neurological impairment and future disability as well as higher mortality [16, 19]. This lack of standardized definitions limits knowledge about response of CNS disease to therapy. Comparability across cohorts is equally limited for this reason. Evaluation for possible CNS involvement in HLH rests on information from three specific areas of investigation: (a) the presence of neurological signs/symptoms, (b) neuroimaging abnormalities, and (c) evaluation of CSF. In Table 1, we outline literature describing the relative frequency of specific findings in the three above-mentioned categories.
Neurological signs/symptoms
Few studies including prospective neurological evaluations of consecutive HLH patients exist [7, 10, 16, 23•]. The findings of these and a number of retrospective efforts have documented the rate of CNS involvement to be in the range of 18–73% [4••, 6, 8, 9, 11••, 24••]. The larger and more systematic of the studies suggest that about 2/3 of all HLH patients (both primary and secondary) experience neurological manifestations [7, 19]. These numbers rest on evaluation of cases that, for the most part, present with systemic HLH and are noted additionally to have CNS features. However, inflammation of the CNS may be the primary and only clinical presentation of HLH [25,25,26,27,28,30]. The prospective treatment study, HLH-2004, will provide some answers to these questions. This study documents standardized neurological outcomes and is due to be published in 2016. Preliminary results suggest frequent neurological involvement in HLH. The study is closed but data is not yet published.
Reported neurological symptoms and/or signs are severe, sometimes life-threatening, and may occur early on in the course of the disease. Seizures are the most common sign of neurological dysfunction, as seen in 33–83% of children who are reported to have CNS-HLH [8, 10, 11••, 16, 19, 24••]. Mental status changes, described variably as irritability, disturbance of consciousness, and encephalopathy also occur commonly (31–47%), suggesting that gray matter dysfunction is relatively common in this population. Additionally, meningism is reported in approximately one third of patients with neurological findings in some cohorts. Focal neurological signs, such as hemiparesis, cranial neuropathies, and ataxia, are seen in 10–20% of reported cohorts [8, 10, 11••, 16, 19, 24••]. However, comparison between cohorts is difficult due to inconsistency in the rates of neurological symptoms reported.
The differential diagnosis for CNS-HLH is broad and includes acute disseminated encephalomyelitis (ADEM), acute necrotizing encephalopathy (ANE), CNS vasculitis, multiple sclerosis, encephalitis, CNS manifestations of rheumatologic disease (such as systemic lupus erythematosus), and other genetically mediated CNS inflammatory disorders such as interferonopathies.
Analysis of CSF
CSF abnormalities are seen in a large proportion of HLH cases with or without neurological symptoms. A lumbar puncture should, therefore, be routinely performed in all children where there is a suspicion of HLH and where no contraindications are present. Analysis of CSF should include standard tests (i.e., cells, protein including fractions, glucose, lactate, and microbiology) and a cytospin with examination for hemophagocytosis.
CSF pleocytosis is seen in 10–47% of HLH patients [7, 10, 11••, 16, 19]. It should be noted, however, that pleocytosis may be a late sign and repeat lumbar punctures may be of value if clinical suspicion remains. Although increased protein levels, as seen in 11–41% of HLH patients [7, 9, 16], are usually only moderately elevated (between 500 and 1000 mg/L, normal range age-dependent 150–400 mg/L), values up to 10,000 mg/L have been reported [10, 31]. Protein levels higher than 2500 mg/L have been associated with stage III abnormalities [4••], but the prognostic value is uncertain as even patients with extremely elevated levels have had a good outcome [31]. High CSF protein levels in an encephalopathic child with unknown diagnosis should raise the suspicion of a neuroinflammatory condition including HLH.
“Abnormal CSF” is defined in many studies as including pleocytosis, increased CSF protein, or both. This classification leads to findings of CSF abnormalities in 16–76% of HLH cases [7, 8, 10, 16]. As a whole, the presence of neurological symptoms and CSF abnormalities of any kind is a negative prognostic marker, e.g., reducing 5-year survival from 67 to 40% [7, 16]. CSF abnormalities may respond rapidly to therapy, with one case series showing resolution of CSF abnormalities in all children within 6 weeks of treatment [10].
Although the pathogenesis of HLH is not fully understood, the clinical symptoms are considered to be mediated by excessive activation of CD8+ T lymphocytes and the release of cytokines such as tumor necrosis factor-α, IL-1β, IL-6, IL-8, and interferon-γ [32,32,33,35]. Limited information from cases reports suggests that other neuroinflammatory markers such as neopterin may be useful for diagnosis of CNS disease [36]. Given similarities with other neurological diseases affecting white matter, biomarkers known to be important for these disorders, e.g., CXCL 13 and neurofilament light chain, should also be studied in the future [37]. Since neurological symptoms may be seen in the presence or absence of elevated CSF protein or pleocytosis [12, 15, 16], finding biomarkers with a higher sensitivity and specificity for CNS HLH would be of great importance.
Hemophagocytosis, which is described to be present in 91% of brain biopsies, mostly located in the meninges [4••] and in 92% of bone marrow samples, was less commonly seen in the CSF (39%) of pediatric cases [7]. Whether the degree of hemophagocytosis in the CSF correlates to the duration and severity of disease, as demonstrated in brain tissue [4••], is not known.
Neuroimaging
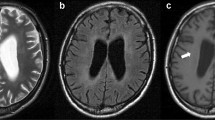
MRI of the brain with gadolinium is the imaging modality of choice in situations in which CNS involvement in HLH is suspected (Figs. 1 and 2). Where MRI is not readily available, CT scans may provide valuable structural and other information, but cannot replace the detailed assessment available from MRI. The range of abnormalities seen on neuroimaging is broad. Descriptive information from retrospective case series including both primary and secondary forms of HLH suggests that multifocal and bilateral abnormalities seen on T2-weighted imaging are almost universally present in primary HLH (89%), with a high rate of symmetric involvement (53%), thus, distinguishing it from ADEM where symmetric involvement is infrequently seen [8]. Furthermore, large, ill-defined, confluent lesions are seen in up to 2/3 of an HLH population [8]. CNS hemorrhage was seen in 5/43 cases in one series of mixed primary and secondary HLH [10]. Chronic changes such as atrophy and calcifications are noted in several series, but information on timing of the imaging and relation to the use of agents such as steroids and in relation to onset of symptoms are not available in these reports [8,8,10, 38]. Administration of contrast is beneficial in this population, with one small case series suggesting the presence of nodular or ring (6/9), and/or leptomeningeal enhancement (5/9) in the majority of cases [38].

Suggested workup for CNS disease in a patient with HLH.

Neuroradiological MRI findings in HLH. a T2w image showing bilateral hyperintense lesions in the cerebellum. b T2w image with hyperintense signal and edema in the left posterior hemisphere and abnormalities in the brainstem. c Diffusion weighted imaging of the same region as in b with lesions imitating cerebral infarction.
Treatment of CNS HLH
Review of the literature
The available literature offers no consensus on treatment directed at CNS involvement in HLH. Evidence-based guidelines are lacking and no clinical trials with a focus specifically on CNS disease have been conducted to date. In order to identify published cases of CNS HLH and its specific treatment, we searched the PubMed and Medline databases using the terms “Hemophagocytic lymphohistiocytosis,” “Familial hemophagocytic lymphohistiocytosis,” or “Macrophage activating syndrome.” The search included studies of both children and adults in the English literature and identified 15 papers focused on treatment of CNS involvement in HLH (summarized in Table 2). Most articles were retrospective case reports or case series.
The majority of published reports describe the use of systemic steroids (primarily dexamethasone) combined with other immunosuppressive therapies (cyclosporine A and etoposide), as in the treatment protocols of HLH-94 and HLH-2004 [7, 15]. Both these protocols used intrathecal treatment weekly during the induction phase from weeks 3–6 in patients who had clinical symptoms of CNS disease progression after 2 weeks of systemic treatment, or in those with worsening or unchanged CSF pleocytosis. The rationale for not using intrathecal chemotherapy in all children with CNS involvement at therapy start was that the CNS symptoms improved with systemic therapy alone in most cases. The HLH-94 protocol [7] used intrathecal methotrexate alone while HLH-2004 [15] used intrathecal prednisolone in addition. Another option for HLH treatment is antithymocyte globulin (ATG rabbit) and methylprednisolone followed by cyclosporine A, which is given until hematopoietic stem cell transplant (HSCT), generally allowing tapering of methylprednisolone [17]. In this protocol, patients with CNS disease also receive intrathecal methotrexate and corticosteroids [17]. As both the definitions of CNS-HLH and the time points of treatment vary, direct comparison between these treatments is difficult. The HLH-94 protocol showed that 81/122 (66%) patients with any neurological involvement at onset of disease were alive and in complete remission at 2 months after start of treatment [7]. This is comparable to the ATG study [17] which stated that in patients with signs of overt neurologic disease the probability of achieving complete remission was 11/19 (58%) within a median time of 1.3 months.
Today, intrathecal methotrexate and corticosteroids (as described in HLH-2004 [15]) has become standard of care in the initial treatment of children with CNS-HLH. However, data on the value of intrathecal therapy in patients with CNS involvement is limited. In the first report on HLH-94, neurological alterations were reported in 35/109 (32%) of the patients at onset. In the 35 affected individuals, symptoms normalized in 21/31 (67%) survivors after 2 months of HLH-94 therapy. The rate of normalization was similar whether intrathecal therapy was used or not as an additional treatment to systemic corticosteroids, etoposide, and cyclosporin (10/15 versus 10/15, respectively) [45]. However, intrathecal methotrexate was not studied in a randomized fashion. Hence, additional studies will be required to better evaluate the value of intrathecal therapy in CNS-HLH.
Interestingly, a single report describes treatment with intrathecal rituximab in post-transplant lymphoproliferative disorder (PTLD) in the CNS, a lymphoproliferative disorder with similar features of CNS-HLH. PTLD is a very rare complication of HSCT. This study described successful treatment of CNS PTLD with intrathecal rituximab therapy in two children who had failed to respond to standard chemotherapy, intravenous rituximab and EBV specific cellular therapy [46]. For patients with HLH secondary to EBV-infection, rituximab has been shown to be beneficial for systemic symptoms [47, 48], but the literature does not provide specific results on CNS disease in these patients.
Alemtuzumab [49], a monoclonal anti-CD52 antibody, is another option that has been used for treatment of HLH [44]. This immunotherapy targets B and T lymphocytes and macrophages. A study on refractory HLH with alemtuzumab has yielded promising results [44], but there was a lack of adequate data to comment fully on the responsiveness of refractory CNS disease. Immunomodulating drugs such as IL-1 receptor antagonists have also been used to treat HLH/MAS with improvements described in case reports [50•, 51] and a case series with 12 patients [52]. However, data on CNS involvement were not available in any of these publications.
Two reports suggest that children with CNS HLH can be cured by initiation of HSCT soon after onset. Successful transplantation may prevent both reactivations and CNS disease progression [39] and may prevent the emergence of neurological late effects [41], though reactivation of CNS disease may occur after HSCT.
Ongoing clinical trials
Hybrid immunotherapy for HLH (HIT-HLH, NCT01104025) and Euro-HIT, two clinical trials utilizing ATG, etoposide, and dexamethasone, recently closed and results are pending. Overall, the rationale for these trials was the potential additive or synergistic effects of combining anti-T cell serotherapy (ATG) with reduced dose-intensity etoposide in order to achieve sustained HLH suppression while minimizing myelosuppression. Because these trials utilize conventional agents with typical IT therapy in the case of CNS disease, it is not expected that they will provide new information regarding the treatment of CNS-HLH.
Another ongoing trial combines alemtuzumab, methylprednisone, and cyclosporine (NCT02472054). The rationale for this trial is based on the T cell depleting effects of alemtuzumab and prior reports of its activity in HLH [44, 53]. The trial is ongoing and results are not available.
Also ongoing is a trial (NCT01818492) testing a new anti-IFNγ monoclonal antibody NI-0501 for the treatment of HLH. This therapy represents a new and targeted approach for treating HLH based on preclinical data from animal models. Although the trial is ongoing and final results are pending, interim results were reported in abstract form (https://ash.confex.com/ash/2015/webprogram/Paper87376.html) in December 2015. This report revealed that three patients with CNS disease were treated and that two were possible to evaluate. Both had resolution of CNS signs and symptoms. At this time, it is not clear how useful NI-0501 will be for CNS-HLH, as results of the trial are still preliminary and these numbers are very small. Of note, a large drug such as an antibody would not ordinarily be expected to cross the blood-brain barrier. However, inflammation is known to open this barrier in other contexts and this also may occur in CNS-HLH.
Another very promising agent is the JAK1/2 inhibitor ruxolitinib shown in a recent study of two murine models of HLH to be effective. In the Rab27a-/- mice, CNS involvement was significantly reduced with ruxolitinib therapy [54]. To our knowledge, there is so far no clinical trial initiated with ruxolitinib.
Treatment considerations—opinion statement
The first step towards optimal treatment of CNS-HLH is prompt and accurate diagnosis. This means looking for CNS involvement even when the patient does not present with obvious signs or symptoms. All patients should receive a brain MRI and lumbar puncture, including assessment of neuro-inflammatory markers if available, as soon as this can safely be performed after diagnosis of HLH. We regard presence of unequivocal neurological symptoms and/or signs, any abnormality in the CSF or brain MRI compatible with an inflammatory process to be consistent with a diagnosis of CNS-HLH. Therapy should be started in all HLH cases with neurological symptoms even if a lumbar puncture or MRI have not been obtained or results are still pending.
Although the optimal treatment for CNS-HLH is unknown, below, we present our approach to therapy, which is based on currently available evidence and/or personal experience. The algorithm in Fig. 1 represents the personal view of the authors how to approach the HLH patient with CNS disease. The treatment suggestions in Fig. 1 include HLH patients with genetic and acquired disease without a known underlying condition such as a malignancy, rheumatologic, or metabolic disease.
A steroid, preferably high-dose dexamethasone, is of importance in CNS-HLH treatment. Results of preclinical studies have shown that dexamethasone has a longer half-life in the CSF and better CSF penetration than does prednisone [55, 56], and in prospective randomized trials, dexamethasone yielded better control of CNS leukemia [57]. The highest standard dose of dexamethasone in the HLH-94/HLH-2004 protocols is 10 mg/m2 per day, but some authors have used 20 mg/m2 per day for limited time periods in patients with severe or refractory CNS-HLH.
Etoposide may be effective in the treatment of CNS-HLH. It has also recently been shown to be effective in a murine model of autoimmune encephalitis by selectively attacking activated T cells [58]. Treatment with etoposide at a reduced dosage (75–100 mg/m2) administered only once a week in combinations with corticosteroids can be used by in patients with MAS-HLH and CNS disease [59].
Our recommendation is that intrathecal Mtx and steroids (as described in HLH-2004) for CNS-HLH should be used as a first-line therapy. Treatment protocols are typically weekly for at least three doses and preferably until all CSF indices and CNS symptoms normalize. Surveillance CSF analyses should be obtained for 2–3 weeks afterwards and later if any symptoms reoccur. Brain MRI’s are typically abnormal for months after resolution of all other aspect of CNS disease so this should not be used in isolation for guiding subsequent therapy, unless clearly indicative of new or worsening problems. A recent case report described the use of intrathecal etoposide as successful salvage treatment for a patient with breast cancer and leptomeningeal metastasis [46]. One author also advocates intravenous thiotepa for refractory CNS-HLH since it readily crosses the blood brain barrier [60] and also leads to high drug levels in the CSF. [61, 62]. However, the use of intrathecal therapy for CNS-HLH is controversial. Because cells infiltrate the brain not only via the meninges but also via vessels, as seen in multiple sclerosis, some argue that invasive intrathecal therapy may not be needed. This is in line with the good response to systemic therapy alone in highly inflammatory CNS conditions other than HLH. There are also risks with intrathecal chemotherapy: neurological adverse effects are well described and may be expected [63]. There are also ongoing concerns regarding intrathecal exposure as a major contributor to CNS late effects in children [64]. However, intrathecal therapy may be necessary in patients in which treatment with dexamethasone, etoposide, or ATG offers good systemic control of HLH, but does not control CNS disease. Future studies are required to understand the value and risks of intrathecal therapy in CNS-HLH.
Even if a patient has responded well to initial therapy of HLH, reactivation of CNS-HLH by the time of HSCT is common. Early transplant in HLH can halt the progression of CNS disease [39, 41]. Therefore, even if HLH is still active, an early transplant should be considered as the risk of late effects is more severe than the risk of transplantation. In patients who have CNS involvement pre-transplant, surveillance LP’s after donor engraftment are advisable to monitor for recurrent/persistent CSF abnormalities. If CNS HLH recurs/worsens after HSCT, as indicated by clinical findings or CSF, additional IT and system therapy should be considered. Finally, all long-term survivors should have longitudinal follow-up for neurological late effects including cognitive and motor evaluations.
Conclusion
CNS-HLH is a life-threatening condition, often but not always associated with systemic HLH, for which appropriate clinical, immunological, and radiological work-up is necessary. Current standard of therapy based on immuno-chemotherapy followed by HSCT in patients with primary disease leads to disease control in most, but not all patients. Novel therapeutic approaches are needed, some of which are currently being explored in ongoing clinical trials.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672–81. Since the first classification of histiocytic disorders in 1987, a number of new findings regarding the cellular origins, molecular pathology, and clinical features of histiocytic disorders have been identified. This revised classification system is based on those findings.
Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015 Feb;62(2):346–352. doi: 10.1002/pbc.25308
Akima M, Sumi SM. Neuropathology of familial erythrophagocytic lymphohistiocytosis: six cases and review of the literature. Hum Pathol. 1984;15:161–8.
•• Henter JI, Nennesmo I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. J Pediatr. 1997;130:358–65. In this paper the currently used staging system for the neuropathologic findings in CNS-HLH is presented.
Trottestam H, Berglof E, Horne A, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. 2012;101:313–8.
Ramachandran B, Balasubramanian S, Abhishek N, Ravikumar KG, Ramanan AV. Profile of hemophagocytic lymphohistiocytosis in children in a tertiary care hospital in India. Indian Pediatr. 2011;48:31–5.
Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118:4577–84.
Deiva K, Mahlaoui N, Beaudonnet F, et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology. 2012;78:1150–6.
Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997;89:794–800.
Yang S, Zhang L, Jia C, Ma H, Henter JI, Shen K. Frequency and development of CNS involvement in Chinese children with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2010;54:408–15.
•• Jovanovic A, Kuzmanovic M, Kravljanac R, et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis: a single-center experience. Pediatr Neurol. 2014;50:233–7. This single centre experience showed that involvement in HLH is common and is associated with poor outcome, highlightning the imoprtande to monitor and treat CNS-HLH.
Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21:245–53.
•• Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr. 2013;163:1253–9. This review presents the advances in genetic and pathophysiologicl research, rapid diagnostic modalities and clinical managment of HLH.
Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44:259–72.
Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
Horne A, Trottestam H, Arico M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140:327–35.
Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120:e622–8.
•• Jackson J, Titman P, Butler S, Bond K, Rao A, Veys P, Chiesa R, Leiper A, Riley L, Gilmour K, et al. Cognitive and psychosocial function post hematopoietic stem cell transplantation in children with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2013;132:889–95.e881–883. This paper presents the first study to have systematically evaluated the cognitive and psychosocial outcomes of HLH survivors. It shows that childhood survivors of HLH are at risk of long-term cognitive and psychosocial difficulties.
Kim MM, Yum MS, Choi HW, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol. 2012;47:273–80.
Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the international registry. FHL study group of the histiocyte society. Leukemia. 1996;10:197–203.
Hirst WJ, Layton DM, Singh S, et al. Haemophagocytic lymphohistiocytosis: experience at two U.K. centres. Br J Haematol. 1994;88:731–9.
Koh KN, Im HJ, Chung NG, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis Working Party. Eur J Haematol. 2015;94:51–9.
• Dao AT, Luong VT, Nguyen TT, et al. Risk factors for early fatal outcomes among children with hemophagocytic lymphohistiocytosis (HLH): a single-institution case-series in Vietnam. Pediatr Hematol Oncol. 2014;31:271–81. Prospective study of 89 children with HLH from a single center in Vietnam.
•• Gratton SM, Powell TR, Theeler BJ, Hawley JS, Amjad FS, Tornatore C. Neurological involvement and characterization in acquired hemophagocytic lymphohistiocytosis in adulthood. J Neurol Sci. 2015;357:136–42. This article is the first report that describes the neurological features of acquired HLH in adults.
Feldmann J, Menasche G, Callebaut I, et al. Severe and progressive encephalitis as a presenting manifestation of a novel missense perforin mutation and impaired cytolytic activity. Blood. 2005;105:2658–63.
Shinoda J, Murase S, Takenaka K, Sakai N. Isolated central nervous system hemophagocytic lymphohistiocytosis: case report. Neurosurgery. 2005;56:187.
Moshous D, Feyen O, Lankisch P, et al. Primary necrotizing lymphocytic central nervous system vasculitis due to perforin deficiency in a four-year-old girl. Arthritis Rheum. 2007;56:995–9.
Rostasy K, Kolb R, Pohl D, et al. CNS disease as the main manifestation of hemophagocytic lymphohistiocytosis in two children. Neuropediatrics. 2004;35:45–9.
Henter JI, Elinder G. Cerebromeningeal haemophagocytic lymphohistiocytosis. Lancet. 1992;339:104–7.
Kieslich M, Vecchi M, Driever PH, Laverda AM, Schwabe D, Jacobi G. Acute encephalopathy as a primary manifestation of haemophagocytic lymphohistiocytosis. Dev Med Child Neurol. 2001;43:555–8.
Voeten M, Maes P, Wojciechowski M, Vandenbossche L, Meyts I, Ceulemans B. Extremely elevated cerebrospinal fluid protein levels in a child with neurologic symptoms: beware of haemophagocytic lymphohistiocytosis. Eur J Paediatr Neurol. 2014;18:427–9.
Henter JI, Andersson B, Elinder G, Jakobson A, Lubeck PO, Soder O. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1996;27:21–5.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166:95–109.
Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. 2013;121:5184–91.
Brisse E, Wouters CH, Matthys P. (2016) Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol 174: 203–17.
Howells DW, Strobel S, Smith I, Levinsky RJ, Hyland K. Central nervous system involvement in the erythrophagocytic disorders of infancy: the role of cerebrospinal fluid neopterins in their differential diagnosis and clinical management. Pediatr Res. 1990;28:116–9.
Kothur K, Wienholt L, Brilot F, Dale RC. CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: a systematic review. Cytokine. 2016;77:227–37.
Goo HW, Weon YC. A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr Radiol. 2007;37:1110–7.
Ouachee-Chardin M, Elie C, de Saint Basile G, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–50.
Tateishi Y, Oda S, Sadahiro T, et al. Continuous hemodiafiltration in the treatment of reactive hemophagocytic syndrome refractory to medical therapy. Transfus Apher Sci. 2009;40:33–40.
Sparber-Sauer M, Honig M, Schulz AS, et al. Patients with early relapse of primary hemophagocytic syndromes or with persistent CNS involvement may benefit from immediate hematopoietic stem cell transplantation. Bone Marrow Transplant. 2009;44:333–8.
Hu Y, Xu J, Wang L, Li J, Qiu H, Zhang S. Treatment of hemophagocytic lymphohistiocytosis with cyclophosphamide, vincristine, and prednisone. Swiss Med Wkly. 2012;142:w13512.
Rajajee S, Ashok I, Manwani N, Rajkumar J, Gowrishankar K, Subbiah E. Profile of hemophagocytic lymphohistiocytosis; efficacy of intravenous immunoglobulin therapy. Indian J Pediatr. 2014;81:1337–41.
Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60:101–9.
Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–73.
Bonney DK, Htwe EE, Turner A, et al. Sustained response to intrathecal rituximab in EBV associated post-transplant lymphoproliferative disease confined to the central nervous system following haematopoietic stem cell transplant. Pediatr Blood Cancer. 2012;58:459–61.
Chellapandian D, Das R, Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013;162:376–82.
Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29:569–73.
Keith MP, Pitchford C, Bernstein WB. Treatment of hemophagocytic lymphohistiocytosis with alemtuzumab in systemic lupus erythematosus. J Clin Rheumatol. 2012;18:134–7.
• Rajasekaran S, Kruse K, Kovey K, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr Crit Care Med. 2014;15:401–8. Retrospective case series of 8 patients in the ICU with secondary HLH. All were treated with anakinra as first line therapy but some also received steroids. Seven out of the eight survived indicating that Anakinra might be initially used for secondary HLH.
Shafferman A, Birmingham JD, Cron RQ. High dose Anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol Online J. 2014;12:26.
Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford). 2011;50:417–9.
Hege K, Quigg T, Delgado D. Alemtuzumab, fludarabine, low-dose TBI, and double umbilical cord transplant for primary graft failure in a patient with recurrent HLH. Pediatr Blood Cancer. 2016;63:361–3.
Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. (2016) Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood 128: 60–71.
Eadie MJ, Brophy TR, Ohlrich G, Tyrer JH. Dexamethasone: pharmacokinetics in neurological patients. Clin Exp Neurol. 1984;20:107–18.
Balis FM, Lester CM, Chrousos GP, Heideman RL, Poplack DG. Differences in cerebrospinal fluid penetration of corticosteroids: possible relationship to the prevention of meningeal leukemia. J Clin Oncol. 1987;5:202–7.
Ito C, Evans WE, McNinch L, et al. Comparative cytotoxicity of dexamethasone and prednisolone in childhood acute lymphoblastic leukemia. J Clin Oncol. 1996;14:2370–6.
McNally JP, Elfers EE, Terrell CE, et al. Eliminating encephalitogenic T cells without undermining protective immunity. J Immunol. 2014;192:73–83.
Palmblad K, Schierbeck H, Sundberg E, et al. High systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol Med. 2014;20:538–47.
Egorin MJ, Akman SR, Gutierrez PL. Plasma pharmacokinetics and tissue distribution of thiotepa in mice. Cancer Treat Rep. 1984;68:1265–8.
Mack F, Baumert BG, Schafer N, et al. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat Rev. 2016;43:83–91.
Heideman RL, Cole DE, Balis F, et al. Phase I and pharmacokinetic evaluation of thiotepa in the cerebrospinal fluid and plasma of pediatric patients: evidence for dose-dependent plasma clearance of thiotepa. Cancer Res. 1989;49:736–41.
Ruggiero A, Conter V, Milani M, et al. Intrathecal chemotherapy with antineoplastic agents in children. Paediatr Drugs. 2001;3:237–46.
Duffner PK, Armstrong FD, Chen L, et al. Neurocognitive and neuroradiologic central nervous system late effects in children treated on pediatric oncology group (POG) P9605 (standard risk) and P9201 (lesser risk) acute lymphoblastic leukemia protocols (ACCL0131): a methotrexate consequence? A report from the children’s oncology group. J Pediatr Hematol Oncol. 2014;36:8–15.
Acknowledgements
The authors would like to thank Carl Gornitzki and Magdalena Svanberg, librarians at the Karolinska Institute University Library, for their assistance with the literature searches. We also would like to thank Dr. Karin Beutel for providing the MRI images.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
AnnaCarin Horne, Ronny Wickström, Michael B. Jordan, Ahmed Naqvi, and Gritta Janka declare that they have no conflicts of interest.
E. Ann Yeh has received personal fees from Novartis and ACI and unrestricted funds for symposium to home institution from Teva.
Jan-Inge Henter has received grants from The Children’s Cancer Foundation of Sweden, The Swedish Research Council, The Swedish Cancer Society, and The Cancer and Allergy Foundation during the conduct of the study; and non-paid consultancy to Novimmune SA.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neurologic Manifestations of Systemic Disease
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Horne, A., Wickström, R., Jordan, M.B. et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis?. Curr Treat Options Neurol 19, 3 (2017). https://doi.org/10.1007/s11940-017-0439-4
Published:
DOI: https://doi.org/10.1007/s11940-017-0439-4