
Abstract
Polymerase Chain Reaction (PCR) is the DNA-equivalent of Gutenberg’s movable type printing, both allowing large-scale replication of a piece of text. De novo DNA synthesis is the DNA-equivalent of mechanical typesetting, both ease the setting of text for replication. What is the DNA-equivalent of the word processor? Biology labs engage daily in DNA processing—the creation of variations and combinations of existing DNA—using a plethora of manual labor-intensive methods such as site-directed mutagenesis, error-prone PCR, assembly PCR, overlap extension PCR, cleavage and ligation, homologous recombination, and others. So far no universal method for DNA processing has been proposed and, consequently, no engineering discipline that could eliminate this manual labor has emerged. Here we present a novel operation on DNA molecules, called Y, which joins two DNA fragments into one, and show that it provides a foundation for DNA processing as it can implement all basic text processing operations on DNA molecules including insert, delete, replace, cut and paste and copy and paste. In addition, complicated DNA processing tasks such as the creation of libraries of DNA variants, chimeras and extensions can be accomplished with DNA processing plans consisting of multiple Y operations, which can be executed automatically under computer control. The resulting DNA processing system, which incorporates our earlier work on recursive DNA composition and error correction, is the first demonstration of a unified approach to DNA synthesis, editing, and library construction.
Similar content being viewed by others


Introduction
While the electronic representation of text in computers allows composing new text and processing an existing piece of text within the same framework, DNA composition and processing are handled completely separately and using unrelated methods. DNA composition, also called de novo DNA synthesis, uses several methods for assembling synthetic oligonucleotides into ever longer pieces of DNA (Stemmer et al. 1995; Merkle 1997; Au et al. 1998; Smith et al. 2003; Xiong et al. 2004; Schatz et al. 2005; Xiong et al. 2006). Much progress has been made in achieving uniform, efficient and automated methods for performing this assembly. DNA processing, on the other hand, has no systematic solution to date, and the various DNA processing tasks are performed by a plethora of manual labor-intensive methods (Kunkel 1985; Wilson 1988; Ho et al. 1989; Landt et al. 1990; Wilson and Murray 1991). Site-directed mutagenesis generates targeted changes including single or few nucleotide insertions, deletions or substitutions, usually via the use of an oligonucleotide primer that introduces the desired modification. These fall into two major categories: those based on primer extension on a plasmid template (Kunkel 1985; Ho et al. 1989; Landt et al. 1990), and PCR-based methods, i.e. overlap extension (Horton et al. 1989; Weiner et al. 1994; Xiao et al. 2007). A common technique is the use of restriction enzymes to cut the DNA molecule at specific sites which enables the joining of DNA fragments that contains matching sites on their ends (Wilson 1988; Wilson and Murray 1991). In vitro homologous recombination methods (Hartley et al. 2000) such as SLIC (Li and Elledge 2007) provide a general method for recombining DNA fragments but are not amenable to recursive composition (Linshiz et al. 2008) as they require cloning of the products at every iteration. Other methods exist which are used to introduce random variation in DNA such as error-prone PCR (Cirino et al. 2003) or random DNA shuffling (Coco et al. 2001). Generally, as most of these methods require iterative steps of mutagenesis, cloning, sequencing and selection, they become inefficient if multiple non-random sequence manipulations are required. Moreover, many of these methods require several steps which are not easily automatable, therefore the time and effort required to create libraries of different mutations scales with the size of the library. Alternatively, these methods impose restrictions on the types of changes which are possible which limit the scope of their usefulness, e.g. restriction enzymes require specific sites to be present. So far no universal method which overcomes these limitations has been proposed and consequently no engineering discipline which eliminates this manual labor has emerged. A general DNA processing method should enable extensive manipulation of a DNA molecule while maximizing the use of existing DNA and minimizing the need for synthesizing new DNA, similarly to the way a text editor enables efficient editing of an existing text, minimizing the need to retype pieces of text that are already available. A general method should also be amenable to full automation and thus enable the creation of large libraries with a small additional effort.
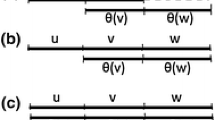
In this work we present a uniform framework for DNA processing that encompasses DNA editing, DNA synthesis, and DNA library construction. The framework is based on one core biochemical operation, called Y, that takes as input two DNA fragments, A and B, and produces the concatenated DNA molecule AB (Fig. 1a). The input fragments A and B can be two individual DNA molecules or two DNA fragments embedded either in one or two longer DNA molecules, and they can be in single strand (ssDNA) or double strand (dsDNA) form. They must, however, be amenable to amplification by a PCR reaction. The output molecule AB of the Y operation is double stranded. This allows the process to be iterated as many times as needed to perform the DNA editing task, as the output of one step is used as the input for the following step. Also, an output of one step can be used as the input for many different processing operations. This property enables the efficient reuse of intermediate DNA fragments.

Y Operation. a Specification of the Y operation, which takes as input two DNA fragments A and B (either ssDNA or dsDNA) that may reside in the same molecule or in different molecules, and produces the concatenated DNA molecule AB. b Implementation of the Y operation (1) fragments A and B are amplified by primers with overhangs to produce overlapping dsDNA molecules marked by phosphate in their 5′ end. Fragments can be the same as their templates or might be a subfragment of it. (2) λ-exonuclease digests the phosphorylated strands resulting in two overlapping ssDNA molecules. (3) Elongation by polymerase in quasi-equilibrium produces the output dsDNA molecule with the concatenated sequence AB
The implementation of the Y operation (Fig. 1b) can be divided into two stages. In the first, each of the two input molecules are amplified from their templates using PCR and extended using primer overhangs, producing two partially overlapping dsDNA molecules, each with a strand marked by an exposed phosphate at its 5’ end. The two marked strands are then enzymatically digested, resulting in two partially overlapping ssDNA molecules. In the second stage, the two overlapping strands form an elongated dsDNA molecule using mutual elongation by DNA polymerase. Unlike the use of restriction enzymes, which require that a specific restriction site be uniquely embedded in the sequence, the elongation step can allow concatenation at almost any location in the sequence as long as the overlapping sequence is sufficiently unique to guarantee specific hybridization.
This work builds upon and extends our earlier work (Linshiz et al. 2008). In our earlier work, the focus was on incorporating error correction in de novo DNA synthesis, and as such offered an improvement over other methods that start with faulty DNA oligonucleotides with the goal of ending with faultless long DNA molecules. In this work the focus is on DNA reuse, and as such it breaks away from existing methods for DNA synthesis rather than tries to improve upon them. Our method offers reuse of DNA when the output molecules needed are similar to an input molecule already available, and also when a library of DNA molecules is needed when library elements have shared components. This forced a change in the basic step; in our previous work each fragment was used once and the overlaps were embedded into the fragments. In our current system each DNA fragment is reused several times and so we add the overlap regions in the primer overhang (through so called “extension PCR”), which could be different for each reuse. A key feature of our basic step, the Y operation, as well as its predecessor (Linshiz et al. 2008), is that its output is of the same type as its input, and therefore it can be used as a basis for recursive composition. In contrast to our previous basic step, where we defined our operation as starting with single strands (oligonucleotides) and ending with single strands, now we start with double stranded DNA (existing material) and end with double stranded DNA. This allows us to start with any material, such as PCR product, a plasmid or annealed oligos.
Results and discussion
The core Y operation, despite its simplicity, allows great flexibility in editing DNA molecules. By applying the Y operation multiple times one can implement all the basic text editing operations on DNA, as demonstrated in Fig. 2.

DNA processing operations. a Simple composition of DNA fragments done by combining Y operations. In addition, simple DNA edit operations can be performed by composing Y operations. b Insertion of an existing DNA fragment into another existing DNA fragment. c Deletion of an internal fragment. d Replacement of an internal fragment by a new fragment. e Cut and Paste in which a fragment is deleted and then inserted in another location. f Copy and Paste, where a fragment B is copied from one location (between A and C) and the copy is inserted in another location (between C and D). g Short insertions or substitutions can be accomplished simply with a single Y operation, as the modified sequence can be embedded in the overlap. h Detailed description of the Copy and Paste operations including primers, their overlap extensions and the required phosphorylation
The “compilation” of a set of Y operations into their biochemical implementation is amenable to various optimizations, similar in nature to those carried out when compiling a high-level computer programming language into machine language. For example, in case of a single or a few codon substitution or insertion, the modified sequence can be embedded in the primer overlap extension (Fig. 2g), resulting in an implementation using only a single Y operation, compared to two Y operations for long insertions (Fig. 2b).
Creating DNA libraries without computer support can be very difficult. Even the creation of a single gene through assembly PCR is very confusing; the abundance of software packages aimed at easing this process is evidence (Xiong et al. 2008). While the basic logic of the Y operation is simple and intuitive, using multi-layered Y operations make the construction even harder as it requires more biochemical steps. In our lab we try to use automation to carry out most of the tasks from designing the construction, through the construction itself and finally cloning and sequencing. A unified approach to DNA processing makes automation much easier and so we find that it has a value in itself.
Planning a DNA processing task
Any desired DNA processing task can be realized by a sequence of the basic edit operations shown in Fig. 2, which can be further decomposed into Y operations. However, such a translation might not yield an optimal editing plan, and therefore we utilize the basic Y operation directly. We developed a Divide and Conquer algorithm to find an optimal set of Y operations to produce the target molecules from the input molecules.
The input to the algorithm is the sequence T of the desired target DNA molecule, as well as a set of sequences S of the available input DNA molecules, which could be naturally available or the result of a previous synthesis or processing task. As output, the algorithm produces a DNA processing plan consisting of a set of Y operations. For a single target molecule, the plan has the form of a binary tree of Y operations, where the leaves are either fragments of the input molecules (with valid PCR primers) or synthetic oligos. Internal nodes correspond to intermediate dsDNA molecules built using Y operations and the root is the target molecule T. If there are multiple target molecules, for example a combinatorial variant library, the plan has the form of a directed acyclic graph in which each internal node has two inputs and one or more outputs. A node with multiple outputs represents a DNA molecule that is used as the source of multiple Y operations. The output of the algorithm includes the list of primers and oligos needed to execute the plan as well. Naturally, the plan need not be executed sequentially: all Y operations at the same level of the tree or graph can be executed in parallel, so the overall time of executing the plan is typically a function of its depth rather than of its size.
For a target molecule, T, the algorithm computes the DNA processing plan as follows. First we identify in T so called “input fragments”, which are maximal fragments in T that occur also in one of the input molecules. Clearly any part in T that does not occur in any input molecule has to be synthesized de novo. The algorithm tries to minimize de novo synthesis by maximizing the use of input fragments in composing T.
Next, all end points of the input fragments and all their midpoints in T are marked. At each recursive application of the planning procedure, the marked target sequence is divided into two adjacent parts at a point selected as follows: All potential division points are sorted according to whether or not they occur in an input fragment. Points which fall between input fragments are preferred division points as they do not disrupt the potential use of an input fragment. The points are further sorted according to their absolute distance from the closest middle point of the neighboring input fragments, as this consideration leads to a balanced division and to better concurrency. In this sorting, points which are at the exact ends of an input fragment are preferred over their close neighbors. This allows maximizing the utilization of input fragments by ensuring that their end points are preferred division point.
Once the candidate division points are sorted from best to worst, the first division point is selected and the algorithm tries to plan a basic step reaction that will combine the two sub-fragments induced by the division point into the target molecule. The necessary primers are planned and validated for specificity, affinity (Tm), dimerization and length constraints for both PCR amplification and elongation reactions of the basic step (See methods). Should a division point not satisfy any of these constraints, it is disqualified and the algorithm tries the next potential division point. If a division point satisfies all chemical constraints, the left and right subfragments of T, Tl and Tr, are considered new targets and the same algorithm is used recursively to plan their construction. Should none of the division points satisfy the chemical constraints the algorithm returns a failure. A division point where either the planning of Tl or Tr fail is also disqualified. The recursive division ends when the target can be extracted from one of the input fragments or when it is small enough to be produced synthetically.
The algorithm produces an efficient DNA processing plan that enables parallel steps on the one hand and makes efficient use of input DNA on the other hand. A more detailed description of the algorithm, in the form of pseudo-code can be found in the SI.
Example 1: implementing all basic editing operations using Y
To demonstrate the implementation of basic editing operations using the Y operation we applied the editing operations depicted in Fig. 2 to a 704 bp molecule containing the wild-type GAL1-10 promoter from S. cerevisiae, resulting in 5 different molecules (See Fig. 3). To demonstrate the replace operation, we used a different DNA molecule taken from the GAL80 promoter. The planning and validation of the chemical constraints were planned as a DNA processing task as explained above. The desired target molecules were constructed in one iteration of the protocol and were confirmed by sequencing.

DNA processing Example 1. a A diagram depicting basic editing operation done on the input molecule NATIVE. Another input molecule, GAL80, is used to demonstrate substitution. Each input is designated a unique color and synthetic parts are shown as red boxes. In the output section, we demonstrate five editing operations. Each colored ellipse designates a part of an input molecule, identified by its color, and the numbers are coordinates inside the input molecule, in bps. Red boxes must be synthesized de novo. b An automatically generated plan to construct our targets consisting of several Y operations (further details in Supplementary Table 1). Each colored shape represents a fragment used in the construction process and these are joined together to create the targets; for the input fragments numbers indicate their coordinates in the input molecule. The heavy arm of the Y operation signifies its left component. We executed this plan with our robot lab automation platform to construct the desired molecules and verified the results by sequencing
Advantages of DNA processing vs. de novo synthesis
In the world of biology, tweaking of genes many times to achieve a desired goal is common practice. As perfect models of biology do not currently exist, most synthetic biological systems are evolved through iterative cycles of design, experimentation, and refinement (Purnick and Weiss 2009). In biological research, some techniques, such as Alanine scanning (Cunningham and Wells 1989), rely on making many different mutations to a gene to study its properties. Both kinds of exploration can benefit from large DNA libraries, i.e. many different but similar variants of the same gene, all of them similar to existing genes. These days de novo synthesis of genes, even at sizes of 5 kbp and more, is a commodity (Gibson et al. 2008). Still, many DNA libraries remain prohibitively expensive. For example, in a 100 variants library we have constructed (see Example 3) 96 variants totaled 29 kbp. Yet, each of the desired genes in this library is a mutation on an existing DNA fragment and using DNA processing, the total number of bases that had to be synthesized de novo (“synthetic fragments”) totaled only 10,600 bases, lowering significantly the cost of raw materials. This is the case in many of our other examples where the amount of synthetic fragments is only a small percentage of the size of the library.
An additional advantage to DNA processing lies in the error rate of resulting constructs. While the error rate of amplifying DNA fragments using PCR is extremely small (around 1 error per 50,000 bp per PCR cycle) the error rate in de novo synthesis is mainly the result of oligo synthesis error rate which is much higher at around 1 error for 160 bp (Hecker and Rill 1998; Tian et al. 2004; Linshiz et al. 2008). Example 1 described in the previous section (target 1) makes a good example. A 704 bp variant synthetically constructed from oligos should contain on average 704/160 = 4.4 errors giving a chance of 1.2% of picking an error-free clone. Reusing existing DNA fragments to create the construct, there are two sources of errors; PCR amplification (two Y operations require two PCR amplification, each of 15 cycles for an average of 1 error in every 1,666 bp or around 0.42 errors per molecule) and the synthetic oligos used in the construction (162 synthetic bases contributing on average 1 error per molecule). Assuming the errors are uncorrelated and are additive every molecule should have on average 1.42 errors giving a chance of 24% of picking an error-free clone. Note that the more synthetic parts a target has, the higher its error rate should be. Mutating a single base in the middle of a gene also forces an increase in error rate as it forces more PCR cycles and the use of two more synthetic oligos in the construction. In a completely synthetic target the error rate should be highest and comparable to de novo synthesis (or slightly higher because of more PCR cycles). In our experience with real world libraries the error rate of DNA processing tasks is low enough so that often a single clone suffices to produce an error-free molecule.
In some cases where molecules contain a large number of synthetic bases or where the construction tree is deep enough the error rate may be too high to isolate an error-free clone. A more favorable approach in this case is identifying error-free fragments and using them in a secondary construction phase to rebuild the molecule with a lower error rate, as was demonstrated in our previous work (Linshiz et al. 2008). Furthermore, with DNA libraries we choose certain nodes which both contain a high error rate and are used many times and those are first constructed, cloned and purified and then used in the construction of the library. This is worthwhile as this eliminated many errors in the final targets in relatively little additional work. A detailed example of this is brought later.
Finally, when constructing DNA libraries it is possible to exploit the sharing of components to speed up the construction of the library. By reusing some components many times the throughput can be increased leading to overall speed up of the construction. We try to optimize the construction of libraries by eliminating nodes whose sequence is contained in other nodes, though this is in no way an optimal solution and the construction of libraries with shared components could benefit from further study.
Further examples of DNA processing tasks
We have already completed several DNA processing tasks using complex sets of Y operations, to serve the needs of fellow scientists and to test and develop our DNA processing platform, and more tasks are in process. Two are presented below, others are shown in the SI, and additional tasks are in process. Example 2 is our first real world task (in collaboration with R. Graef). It demonstrates the ability of our Divide & Conquer algorithm to reuse input fragments and intermediate products. Example 4 (see SI) exemplifies shuffling parts of genes together.
Example 2: editing a wild-type gene into a library of combinatorial variants to aid protein design
We used a wild-type gene, DRI, which is 813 bp long, which has two variants with known binding affinities but a third, intermediate, affinity is desired. The 12 mutations separating the two variants were mapped and ranked according to the likely effect on affinity. A library of 11 variants was designed with each mutant changing one of the amino acids with the aim of optimizing the binding affinity of the protein. This kind of designed library allows one to scan for a desired property of a protein with higher chances of success than simple random mutagenesis and with fewer variants. Figure 4a illustrates the desired sequences as a function of the input gene, each with 2–12 amino acid substitutions. The construction plan (Fig. 4b) is 5 levels deep and demonstrates both efficient utilization of the input DNA molecule as well as the sharing of library components among different variants. The library was constructed using our automated platform.

DNA processing Example 2. a 11 variants of a protein of 271 amino acids. Each variant contains 2–12 amino acid substitutions. Coordinates are not shown where a fragment is too small. b The DNA processing plan demonstrates the efficient reuse of DNA fragments to build the entire library. Each non-target node is depicted as part of a target to which it contributes. c Further compactization of the plan is achieved by eliminating internal nodes with sequences contained in other nodes, as often occurs in combinatorial libraries. d A detailed graph describes the protocol reactions composition including primers. This complex graph is translated into a robot control program that performs the specified plan. In reality, each arrow in the graph is translated into one or more reagent transfers in the robot control program
The compositionality of the Y operation
One basic property of the Y operation is that it is compositional. That is, it is possible to directly use the output of one Y operation as the input of another Y operation, allowing the composition of several Y operations to create a complex product. Even though theoretically this is possible with many methods, purity of the products might limit this in practice. That is, the concentration of the desired product might be low either because some of the reagents did not react or because non-specific products are created in the process and require additional expensive purification steps in order to be fed into the next iteration. All cloning and in vitro recombination methods have limited efficiency, that is only a small fraction of the reactants react to create ligated products (Aslanidis and de Jong 1990) and therefore must be transformed into bacteria under selective pressure to produce the desired output. In a newly developed method (Wang et al. 2009) the purity is high enough (around 30% transformed cells) that several transformations have been done consecutively with sequencing and searching happening only after the last transformation. A rather large body of literature describes the construction of genes through different overlap extension methods. So called two-step methods (such as PCR-based two-step synthesis (Xiong et al. 2004)) actually use the output of the first step as input for the second step. We are not aware of attempts to use overlap extension more than twice consecutively but in most works the output of one step contains nonspecific products (An et al. 2005; Xiao et al. 2007) and the desired product is isolated through cloning. In our experience this kind of nonspecific products would hinder the compositionality of the operation. On the other hand, we have been successful in applying our method five times consecutively (see Example 2), each time taking the product of one step to be the input of the following step.
The fact that our Y operation can be used iteratively is useful in several ways; it allows the building of large and complicated DNA molecules. It allows construction in trees, allowing parallelism in construction which decreases construction time. lastly, with Y, the output can be used as the input for several Y’s and thus allows the reuse of created fragments. A strategy we can take while building complicated libraries is the deliberate purification of intermediate targets, that is, instead of building the entire library in one sweep, we may choose several nodes, purify them (for example, by cloning and sequencing) and use them afterwards for the rest of the construction. We do this when the cost of the additional purification step is less than the additional cost of the errors that will be introduced by these intermediate nodes. For example, in Example 4, we calculated that an intermediate node that is 272 bp long should have an error rate around 0.34 errors/molecule or around 0.00125 errors/bp (taking into account both oligos and polymerase errors). This node was subsequently used to construct 10 different targets and would have contributed roughly 3.4 errors to the entire project. We decided that the cost of purifying this node (cloning and isolating an error-free clone) would be cheaper than eliminating those errors in the end despite the fact that it would add additional steps to the project. Similarly, three other nodes were also purified during the project to reduce overall error rate. In any case, for every target we calculate our expected error rate and predict the amount of clones needed of each to reach a target with no errors. In any case when this number is too high, and in cases where some nodes are reused many times, they can be purified before being used.
Note that while the Y operation superficially resembles other overlap extension methods (Horton et al. 1989; Weiner et al. 1994; Xiao et al. 2007) a key difference is the creation of single strands through enzyme digestion in our method allowing elongation to occur in equilibrium, this in turn allows specific and complete elongation of the reactants and, if their concentration is equal, ensures that there are no undesired products or left-over reactants.
Example 3: promoter variants library
In another example (in collaboration with E. Segal), we study a yeast promoter in detail. Ninety-six different variants of the native promoter were designed and constructed. The constructs range in size from 577 to 721 bp with at least the first 220 bp and the last 70 bp common to all constructs (Fig. 5a). The total size of the library (minus the parts identical in all variants) is approx. 29 kbp. The variants share many features which allow for effective reuse of fragments in the construction (Fig. 5b). In addition, each target of this library was used as input for one common additional Y operation (not shown). In this Y operation a 1,600 bp gene used for selection was added before the construct as well as short recombination sequences. This brings the maximum depth of the library to seven and the size of each of the final constructs to ~2 kbp The products of this final Y operation were used directly to transform yeast. Each of these constructs was built once and for each, between 1 and 4 clones (average 2.1 clones/construct) were sequenced. Out of 96 constructs, 61 were found error-free in at least one of the clones and 16 were found containing an error in a non-important area, for a total of 77 successfully built constructs. When we investigated the 19 failures, most were found to contain sequences that are known to be hard to amplify by PCR such as large palindromic sequences (17 bp) and long poly-A sequences, stressing both the crucial role of PCR in our method and the robustness of our method in cases where amplification by PCR is not a problem. We believe that in the future such failures will be minimized by computationally predicting which of the sequences are hard to amplify.

DNA processing Example 3. a Diagram of the different variants in a 96 variants library. Blue signifies parts of the wild-type promoter, brown—parts of a plasmid used in the construction. Red signifies parts which are completely synthetic or too short to be used as a part of available fragments and so must be synthesized anew. Note the graphics do not show the first 200 bp which are identical in all constructs. b A diagram of the plan of the construction shown without labels. Each purple square is a target; a brown square is an available fragment and a green square signifies an oligo (synthetic fragment). Blue squares are intermediates of the process (Color figure online)
A DNA processing system
The basic Y operation, which is amenable to automation, can be combined with the Divide and Conquer planning algorithm described above into a fully automated DNA processing system. The system receives a set of DNA molecules as input as well as the specification of one or several desired targets. The planning algorithm computes a plan to construct the target molecules, which is then translated into a robot control program that implements the plan. Given the input molecules and the requisite oligos, primers and reagents, the control program instructs the robot to perform the sequence of Y operations that produce the target molecules. The final targets can then be cloned and sequenced to find a correct target molecule. Alternatively, a two stage error correction step can be used. In the first stage each target is cloned in vitro using single-molecule PCR and sequenced (Ben Yehezkel et al. 2008). Error-free fragments are then identified and used to rebuild the same targets, using the same plan, resulting in a lower error rate (Linshiz et al. 2008). These rebuilt fragments are then cloned in vivo and sequenced to find an error-free target molecule.
The need for sophisticated processing of DNA is ever increasing. As in software and hardware engineering, the development of new genes and biological systems is iterative in nature and requires the ability to quickly modify an existing design and test the results.
In this work we have presented a DNA processing system that uses a computational algorithm combined with biochemical protocols and robotic automation to enable flexible and efficient DNA processing while maximizing the use of existing DNA molecules and shared components. The main limitations of our approach are derived from the use of PCR, which means that DNA molecules that are very long and/or are of very low complexity cannot be processed effectively. Still, our method supports a broad range of editing operation on DNA molecules and allows the use of most sequences as input and output of DNA processing. This platform may be used in the future as a powerful tool for constructing genetic circuits from an existing repertoire of genes and control regions.
References
An Y, Ji J, Wu W, Lv A, Huang R, Wei Y (2005) A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl Microbiol Biotechnol 68(6):774–778
Aslanidis C, de Jong PJ (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18(20):6069–6074
Au L, Yang F, Yang W, Lo S, Kao C (1998) Gene synthesis by a LCR-based approach: high-level production of leptin-L54 using synthetic gene in Escherichia coli. Biochem Biophys Res Commun 248(1):200–203
Ben Yehezkel T, Linshiz G, Buaron H, Kaplan S, Shabi U, Shapiro E (2008) De novo DNA synthesis using single molecule PCR. Nucleic Acids Res 36(17):e107
Cirino P, Mayer K, Umeno D (2003) Generating mutant libraries using error-prone PCR. Methods Mol Biol 231:3–9
Coco W, Levinson W et al (2001) DNA shuffling method for generating highly recombined genes and evolved enzymes. Nat Biotechnol 19(4):354–359
Cunningham BC, Wells JA (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244(4908):1081–1085
Gibson DG, Benders GA et al (2008) Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319(5867):1215–1220
Hartley J, Temple G, Brasch M (2000) DNA cloning using in vitro site-specific recombination. Genome Res 10(11):1788–1795
Hecker K, Rill R (1998) Error analysis of chemically synthesized polynucleotides. Biotechniques 24(2):256–260
Ho S, Hunt H, Horton R, Pullen J, Pease L (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77(1):51–59
Horton R, Hunt H, Ho S, Pullen J, Pease L (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77(1):61–68
Kunkel T (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA 82(2):488–492
Landt O, Grunert H, Hahn U (1990) A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene 96(1):125–128
Li M, Elledge S (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4(3):251–256
Linshiz G, Yehezkel T et al (2008) Recursive construction of perfect DNA molecules from imperfect oligonucleotides. Mol Syst Biol 4:191
Merkle RC (1997) Convergent assembly. Nanotechnology 8:18–22
Purnick PE, Weiss R (2009) The second wave of synthetic biology: from modules to systems. Nat Rev Mol Cell Biol 10(6):410–422
Schatz DO, Dr. Schwer H, Dr. Horn G (2005) De novo enzymatic production of nucleic acid molecules. Sloning Biotechnology GMBH (DE)
Smith H, Cr Hutchison, Pfannkoch C, Venter J (2003) Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc Natl Acad Sci USA 100(26):15440–15445
Stemmer W, Crameri A, Ha K, Brennan T, Heyneker H (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164(1):49–53
Tian J, Gong H et al (2004) Accurate multiplex gene synthesis from programmable DNA microchips. Nature 432(7020):1050–1054
Wang HH, Isaacs FJ et al (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460(7257):894–898
Weiner M, Costa G, Schoettlin W, Cline J, Mathur E, Bauer J (1994) Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 151(1–2):119–123
Wilson G (1988) Cloned restriction-modification systems–a review. Gene 74(1):281–289
Wilson G, Murray N (1991) Restriction and modification systems. Annu Rev Genet 25:585–627
Xiao YH, Yin MH, Hou L, Luo M, Pei Y (2007) Asymmetric overlap extension PCR method bypassing intermediate purification and the amplification of wild-type template in site-directed mutagenesis. Biotechnol Lett 29(6):925–930
Xiong A, Yao Q et al (2004) A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res 32(12):e98
Xiong A, Yao Q et al (2006) PCR-based accurate synthesis of long DNA sequences. Nat Protoc 1(2):791–797
Xiong AS, Peng RH et al (2008) Chemical gene synthesis: strategies, softwares, error corrections, and applications. FEMS Microbiol Rev 32(3):522–540
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Uri Shabi and Shai Kaplan are Equally Contributed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shabi, U., Kaplan, S., Linshiz, G. et al. Processing DNA molecules as text. Syst Synth Biol 4, 227–236 (2010). https://doi.org/10.1007/s11693-010-9059-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11693-010-9059-y