
Abstract
Carbothermic smelting of ores to produce metals or alloys in alternating current open/semiclosed and closed submerged arc furnaces, or in closed direct current furnaces, results in large volumes of CO-rich off-gas being generated. Most of the CO-rich off-gas is cleaned and flared on stacks, since the storing of large volumes is problematic due to the associated toxic and explosive risks. Flaring of CO-rich off-gas results in significant wastage of energy. In this study, an alternative method to partially capture the thermal energy associated with off-gas combustion, in the form of silicon carbide (SiC) generated from waste materials (quartz and anthracite fines), is proposed. SiC can partially replace conventional carbonaceous reductants used to produce alloys such as ferrochromium. The influences of quartz and anthracite particle size, treatment temperature, and gaseous atmosphere (nitrogen or air) on SiC formation were investigated. A quartz-anthracite mixture with 90 pct of the particles < 350.9 µm carbothermically treated at 1873.15 K (1600 °C) resulted in almost complete conversion of quartz to SiC in both nitrogen and air atmospheres. The study indicated significant potential for industrial application of the process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pyrometallurgical smelting of ores is used to produce various metals and alloys. In this paper, ferrochromium (FeCr) will be considered as an example. FeCr, a crude alloy consisting principally of chromium (Cr) and iron (Fe), is produced by the energy-intensive carbothermic reduction of chromite (formula [(Mg,Fe2+)(Al,Cr,Fe3+)2O4]).[1] FeCr is mainly produced using open/semiclosed and closed submerged arc furnaces (SAFs), or direct current (DC) furnaces,[2,3,4] and is primarily used as a source of new Cr units in stainless steel production.[5]
Four prominent waste streams are generated during FeCr production, i.e., slag, CO-rich off-gas, bag filter dust produced during the cleaning of open/semiclosed SAF off-gas, and sludge from wet scrubbing of closed SAF or DC arc furnace off-gas.[4,6] Each of these waste streams is handled and/or treated separately. Any remaining FeCr is usually removed from slag by magnetic separation or jigging, after which the slag is either stockpiled or utilized in various commercial applications.[7,8,9] Bag filter dust and sludge from the wet scrubbing system are usually stored in fit-for-purpose landfill sites, after being treated to remove hexavalent Cr, if required.[2,10,11,12] CO-rich off-gas percolating through the feed material bed of open/semiclosed SAFs ignites as it reaches the top of the bed, due to air ingress into these types of furnaces. Thereafter, the combusted off-gas is extracted and cleaned with bag filters before being released into the atmosphere.[2,12,13] Of specific relevance in this study is the CO-rich off-gas originating from closed SAFs or DC furnaces, which does not combust on the furnace feed material bed, due to the closed nature of these furnaces.
Limited information regarding FeCr off-gas composition is available in the peer-reviewed public domain. Niemelä et al.[14] and Kapure et al.[15] indicated that closed FeCr SAF off-gas typically consists of 60 to 90 pct CO, 10 to 40 pct CO2, 2 to 7 pct N2, and 2 to 10 pct H2. Schubert and Gottschling[16] reported the gas composition of a FeCr DC furnace as 58 to 64 pct CO, 2 to 6 pct CO2, 26 to 34 pct H2, 0 to 5 pct N2, and < 1 pct O2. The furnace technology and operating conditions utilized will obviously affect the gas composition. Typically, it can be assumed that the CO2 content of SAF off-gas will be higher than that of DC furnace off-gas. This is due to additional air ingress allowed by SAF operators, which raises the temperature in the freeboard (area between the feed material bed and furnace roof), resulting in improved Søderberg electrode baking.[17] Obviously, such electrode baking is irrelevant for the graphite/prebaked electrodes that are commonly used in DC furnaces,[4] and the freeboard temperature with the DC open arc is already very high. In contrast, H2 off-gas content is typically lower for SAFs than for DC furnaces, since the feed materials for SAFs are commonly preheated[14] or prereduced,[18] while DC furnace feeds are currently not. Also, higher freeboard DC temperatures result in more effective water to H2 conversion.
Between 30 and 35 pct of the cleaned CO-rich off-gas is typically utilized as an on-site energy source for, e.g., heating ladles, raw material drying, preheating the furnace charge, and chromite pellet sintering/prereduction.[14] Unused cleaned off-gas is typically flared on top of purpose-designed stacks.[19] The main reasons why CO-rich off-gas is usually not stored in large volumes on-site are its toxicity to humans via inhalation[20] and explosive risk.[14] However, off-gas flaring results in an energy loss of 1.8 to 2.4 MWh/t FeCr produced, assuming that 650 to 750 Nm3 CO is typically generated per ton FeCr[14] and the energy content is 2.8 to 3.2 kWh/Nm3 CO gas.[14] Considering that the specific electricity consumption (SEC) for closed FeCr furnaces (SAFs and DC furnaces) varies between 2.4 and 4.2 MWh/t FeCr,[5] the aforementioned energy loss is very significant.
Due to increased pressure on profitability, (e.g., due to increasing electricity costs) and increased environmental concerns, (e.g., reduction in carbon footprint and carbon tax), methods have been developed in an attempt to utilize CO-rich off-gas. One such method is the combustion of cleaned off-gas in so-called co-gen sets, which are internal combustion engines utilizing CO- and H2-containing off-gas as a fuel source to drive alternators for producing electrical power.[16] However, these engines are susceptible to breakdowns caused by the fluctuating energy contents associated with varying CO and H2 contents in off-gas, which significantly reduces their operational life-spans. In addition, solid matter present in off-gas may result in internal abrasion of engine components.[16] Another method of utilizing CO-rich off-gas is combustion to produce steam for steam turbine electricity generation.
In this research, the authors investigated fundamental aspects associated with an alternative approach to utilize CO-rich off-gas. It is suggested that the thermal energy associated with the combustion of such off-gas can at least partially be stored in the form of chemical energy, by production of silicon carbide (SiC) from waste materials generated on-site. Saddow and Agarwal[21] presented a general review of SiC associated production processes. Typically, SiC is commercially produced by Acheson- or Lely-type processes. These processes are based on the sublimation of silicon dioxide (SiO2) and its reaction with a carbonaceous material, which requires very high process temperatures. Additional relevant literature is considered in Section II (Materials and Methods), since such work guided the experimental conditions considered.
Traditionally, SiC is used as a ceramic material and is characterized as an extremely hard and tough compound, possessing high chemical and thermal stability.[22] However, it has also been reported that SiC can be used as a partial replacement for conventional carbonaceous reductants, (e.g., coke, char, and anthracite), to produce alloys such as FeCr,[23,24] as well as platinum group metals (PGMs).[25] One of the lead authors of this paper was also involved in a trial during which SiC was used as a partial replacement for carbonaceous reductants on a large commercial open/semiclosed FeCr SAF. During this trial, significant increases in FeCr production volumes were observed. However, after completing the trial, continuous operation using SiC as a partial replacement for carbonaceous reductants was not implemented, due to the high unit cost of commercially available SiC. The use of SiC as a reductant during FeCr production results in the SiC being oxidized to form SiO2, while the oxides in the ores are reduced, as illustrated in Reactions [1] and [2].
Materials and Methods
Materials
Materials used in this study, i.e., quartz and anthracite fines, were obtained from a large FeCr producer that uses these materials in FeCr production. FeCr producers utilizing closed SAFs minimize the consumption of fine feed materials to prevent furnace bed sintering, which may lead to gas eruptions and dangerous bed turnovers.[13] The quartz and anthracite fines were screened out from lumpy anthracite (serving as a reductant) and lumpy quartz (serving as a flux) on site by the FeCr producer. Such screening is common for FeCr producers.[3,5] These materials can therefore, at least partially, be classified as waste materials, although the fine anthracite could be included as a carbon source in oxidative sintered pellets that are produced on site.[26] The fine quartz could be used in sand molds for casting liquid FeCr to produce FeCr ingots (large dome-shaped FeCr metal blocks); however, the fine quartz is typically not fine enough to have the required binding characteristics. Fine quartz could also be used as fine aggregate in concrete; however, the need for the last application on site is limited.
The fine quartz was characterized using quantitative X-ray diffraction (XRD) analysis (described in Section II–D). It revealed a composition of 94.9 pct SiO2, 2.7 pct illite (K0.65Al2[Al0.65Si3.35O10](OH)2), and 2.4 pct magnesioferrite (Fe2MgO4). A detailed characterization of the fine anthracite used in this study was previously presented by Kleynhans et al.[18] and is therefore not repeated in detail here. Of importance for this study were the fixed carbon content of the anthracite (75.1 pct) and the volatile content (6.9 pct). The fixed carbon content was required, since the C (not anthracite) input into the SiC formation reaction needed to be known. When heated, the volatile compounds in the anthracite decompose and are released as in-situ-formed gas that contain H2.[27] Li et al.[28] indicated that the reduction of quartz to SiC is accelerated by in-situ-formed CH4, which is generated by the reaction between C and H2. Thus, although other screened-out fine reductants may be available at a typical FeCr producer, only anthracite was considered as a reductant in this study, since its volatile content is usually higher than that of coke (the most common reductant used in FeCr smelting) and it is generally much less expensive than coke.
Sizing of Quartz and Anthracite
Since particle size was one of the parameters considered in this study, it was decided to prepare samples by two different methods, i.e. size partitioning and milling. Size partitioning of the as-received material was conducted by screening it into several size fractions, i.e., < 106, 106 to 150, 150 to 250, and 250 to 500 µm. Screening was performed using a Haver EML Digital Plus shaker and Haver & Boecker sieves. During milling, 2 g of quartz and 6 g of anthracite was milled together for various times, i.e., 10, 20, 30, and 60 seconds. A Siebtechnik pulverizer was used for milling purposes. All parts of the pulverizer that were in contact with the mixtures were made of tungsten carbide to minimize possible Fe contamination,[18,26] as Fe can promote SiC and silicon nitride (Si3N4) formation.[29,30,31] Laser diffraction particle sizing using a Malvern Mastersizer 2000 was used to determine particle size distribution. In order to avoid the use of a chemical dispersant, samples were ultrasonicated prior to the particle size measurements. Mechanical stirring was set to 2000 rpm, and laser obscuration was kept between 10 and 15 pct.
The equivalent particle sizes (in µm) at which 90 pct of the particles are finer (d90), as well as d50 and d10 (both defined in a similar manner as d90) of the screened size fractions and milled mixtures, as described in the previous paragraph, are presented in Table I.
Experimental Setup for SiC Formation
The mixing ratio of all carbothermically treated (defined as high-temperature treatment in the presence of a carbonaceous reductant) quartz and anthracite mixtures, obtained either from mixing size-fractioned anthracite and quartz, or from co-milled quartz and anthracite (Section II–B) was kept constant at a C/SiO2 molar ratio of 11.86. This equated to 60 g anthracite (45 g C, according to the fixed carbon content determined by proximate analysis) and 20 g quartz (18.98 g SiO2, according to the SiO2 content determined by Rietveld-refined XRD analysis) being used in each experiment. This C/SiO2 molar ratio is significantly higher than the required stoichiometric ratio of 3, according to Reaction [3] representing the overall reduction reaction[28,32,33]:
The ∆G°, as well as all such further values presented in this paper, was determined by the thermodynamic modeling software package HSC.[19,34] The abovementioned fixed C/SiO2 molar ratio was not determined experimentally, but selected based on previous findings and consideration of the possible industrial application. Considering previous findings, Agarwal and Pad[35] determined that a C/SiO2 molar ratio > 5 would be required for very fine milled material (particle sizes < 75 and < 20 µm, for C and SiO2, respectively) for SiC formation. However, since our mixtures were significantly coarser, a much larger C/SiO2 molar ratio was selected, as recommended by Agarwal and Pad.[35] Considering the possible industrial application, carbonaceous reductant cost is generally considered as the joint second largest cost component in FeCr production.[36] Since fine C reductants are significantly cheaper than lumpy C reductants, excess (unreacted) C reductant was not considered as negative in this investigation, since uncreated C could still serve as a reductant in the FeCr smelting process, while lowering production cost by effectively replacing lumpy reductant. In contrast, excess SiO2, or low C/SiO2 molar ratios, will serve little cost benefit in the possible industrial application.
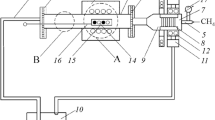
Carbothermic treatment was performed in an 18-kW Lenton Elite tube furnace with a Schunk AluSIK type C610A impervious mullite ceramic tube (75 mm × 65 mm × 1500 mm) with a chemical composition of 60 pct Al2O3 and 40 pct SiO2. Figure 1 presents a simple schematic illustration of the furnace setup used.

Schematic illustration of the furnace setup used
As shown in Figure 1, ceramic heat shields were inserted at both ends of the furnace tube to increase the length of tube that could maintain a stable working temperature. In addition, these heat shields protected the stainless steel caps fitted to seal the furnace tube ends. The stainless steel caps had a gas inlet at one end of the furnace tube, and a gas outlet at the other. The thermocouple used for temperature measurements was located at the center of the stable workable temperature zone. The furnace was heated by twelve electrodes, which were located adjacent to the furnace tube. Samples were placed in ceramic boats and placed within the stable temperature zone.
Li et al.[28] investigated the effects of argon (Ar) and hydrogen (H2) atmospheres on the carbothermic production of SiC from quartz and found that H2 was a better (more productive) atmosphere than Ar. However, considering the nature of the suggested method for producing SiC using combustion of CO-rich off-gas as the heat source, such atmospheres will not be realistic, or economically feasible. The intended combustion of CO-rich off-gas will require air as an oxidizer, which will result in large volumes of nitrogen (N2) being introduced into the process (since normal atmosphere contains > 78 pct N2). Therefore, during this study carbothermic quartz reduction was mainly investigated in a N2 atmosphere, with some CO2 and CO being generated in situ. The N2 atmosphere was maintained within the furnace tube by utilizing a N2 flow rate of 1 NL min−1 during all experiments. Prior to each experiment the furnace tube, already loaded with samples, was flushed with N2 for 30 minutes at a 2 NL min−1 flow rate. During carbothermic treatment, the furnace temperature was increased at a rate of 17.8 K min−1 until the designated temperature was reached. Samples were exposed to the designated maximum temperature for 120 minutes. The furnace was then turned off and allowed to cool to room temperature, while a 1 NL min−1 flow of N2 was maintained with the samples in the furnace tube. The samples were then collected and stored in an airtight container until further analysis.
The temperature range investigated also had to be established. Since the formation of SiC from quartz is an energy-intensive reaction (Reaction [3]), it will require a large amount of heat from CO-rich off-gas combustion. The ∆G° for Reaction [3] will reach equilibrium at approximately 1813.15 K (1540 °C) (determined with HSC). According to Niemelä et al.,[14] pure CO gas can burn in air at 2523.15 K (2250 °C). This value was confirmed by Du Preez et al.[19] using the thermodynamic software program HSC to determine the adiabatic flame temperature of pure CO combustion.[19,34] The aforementioned authors further determined that a gas composition of 60 pct CO and 40 pct CO2, which is at the low end of the CO composition of CO-rich off-gas,[14,15,16] will burn at an adiabatic flame temperature of approximately 2063.15 K (1790 °C). Therefore, the temperatures that could be attained by CO-rich off-gas combustion will be sufficient to sustain, or even surpass, the temperature required for SiC formation. Considering the aforementioned, the temperature range of 1673.15 to 1873.15 K (1400 to 1600 °C) was investigated. This range is much lower that the temperatures typically achieved in the Acheson- or Lely-type processes for commercial production of SiC and is similar to the temperature range proposed by Moshtaghioun et al.[37] of 1473.15 to 1723.15 K (1200 to 1450 °C) for microwave production of SiC. For the formation of Si metal from SiO2 and C, much higher temperatures would be required;[38] however, Si metal formation is beyond the scope of the current paper.
Scanning Electron Microscopy and X-ray Diffraction Analysis
Scanning electron microscopy (SEM) with energy dispersive X-ray spectroscopy (EDS) was used to perform surface characterization of carbothermic-treated samples. A FEI Quanta 200 scanning electron microscope with an integrated Oxford Instruments INCA 200 energy dispersive X-ray spectroscopy microanalysis system was used. Samples were prepared in two different manners prior to SEM analysis. Firstly, samples were mounted onto an aluminum specimen stub with adhesive carbon-coated tape and subsequently coated with a thin layer (approximate 5 nm) of gold-palladium in order to determine the particle morphologies. Second, in order to analyze the surface and subsurface chemical compositions, the samples were set in a carbon-based resin, polished, and coated with gold-palladium before the SEM-EDS analyses were conducted.
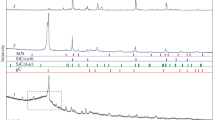
Phase analysis of bulk samples (overall sample) was performed by XRD using a Röntgen diffraction system (PW3040/60 X’Pert Pro) and a back-loading preparation method to determine the crystalline phases and percentages thereof. The samples were scanned using X-rays generated by a copper (Cu) Ka X-ray tube. Measurements were carried out between variable divergences and fixed receiving slits. Phases were identified using X’PertHighscore Plus software. The relative abundances of the detected phases were refined using the Rietveld method.[39] Figure 2 indicates an example of observed and calculated XRD patterns, as well as the difference between the two patterns. The goodness-of-fit (GoF) value for the presented pattern was 1.91. The GoF value for all quantitative Rietveld-refined XRD results presented in this study was ≤ 2.04.

An example of a Rietveld-refined XRD pattern of fine quartz carbothermically treated with anthracite
TG Analysis
Thermogravimetric (TG) analysis was performed using a Netzsch Model STA 449 F3 instrument with a high-temperature rhodium furnace. Approximately 0.5 mg of powdered sample was placed in the manufacture’s Al2O3 crucible and enclosed using the crucible lid. Samples were then heated at a rate of 30 K min−1 to 1873.15 K (1600 °C), then allowed to cool to room temperature. A N2 atmosphere was maintained within the furnace chamber by continuously purging the chamber with N2 at a flow rate of 20 mL min−1. All TG results reported were corrected by a baseline obtained under identical conditions using an empty crucible.
Results and Discussion
Effect of Particle Size
For a constant weight of particulate matter, a decrease in the particle size will result in an increase in surface area. Therefore, the intimate contact between quartz and anthracite particles will increase as a function of the decreasing particle size. Intimate particle contact, promoted by a finer particle size, might be important to promote SiC formation, since solid-state reactions between SiO2 and C (Reaction [4]) are involved in the formation of SiC (Reactions [4] and [5])[32,40]:
Reaction [4] presents the endothermic initial solid-state reaction between SiO2 and C, producing SiO(g) and CO(g). Subsequently, the volatile SiO(g) reacts with solid carbon in a CO/CO2 atmosphere to produce SiC(s) and CO(g) according to the exothermic Reaction [5].
To investigate the effects of quartz and anthracite particle sizes, the finest quartz size fraction (d90 of 119.5 µm, Table I) was carbothermically treated with the increasingly larger anthracite particle size fractions (d90 of 105.3 to 682.6 µm, Table I) at the maximum investigated temperature 1873.15 K (1600 °C), as indicated in Figure 3(a). Subsequently, the finest anthracite size fraction (d90 of 105.3 µm, Table I) was carbothermically treated at 1873.15 K (1600 °C) with the increasingly larger quartz particle size fractions (d90 of 119.5 to 556.8 µm, Table I) (Figure 3(b)). These and subsequent results only indicate the Si-containing compounds normalized to 100 pct (i.e., Si-containing compounds total 100 pct). Non Si-containing compounds such as unreacted C, as well as other compounds that could be detected with the XRD method used (Section II–D), such as MgAl2O4 and AlN formed from impurities in the quartz and anthracite, are not indicated. Also, since SiC can occur in various forms, it is important to specify which form was observed. All XRD diffraction patterns (an example of which is given in Figure 2) indicated that the formed SiC occurred as cubic-SiC (β-SiC). This observation is in agreement with results published by Muranaka et al.,[41] which indicated that β-SiC primarily forms at temperatures < 1973.15 K (< 1700 °C).

Average percentage (pct) composition and standard deviation (based on three experimental repeats) of the finest quartz size fraction (d90 of 119.5 µm, Table I) carbothermically treated with the increasingly larger anthracite particle sizes fractions (a), as well as, the finest anthracite size fraction carbothermically treated with the increasingly larger quartz particle size fractions (b). Both sets of experiment were conducted at a maximum temperature at 1873.15 K (1600 °C)
The results presented in Figures 3(a) and (b) indicate that not only was SiC formed, but also Si3N4. This was expected, since carbothermic reduction of SiO2 in the presence of N2 result in Si3N4 formation[42]:
According to the ∆G° of Reaction [6] (determined with HSC), the reaction will initiate at a temperature of approximately 1823.15 K (1550 °C), which is lower than the actual maximum temperature at which these tests were conducted 1873.15 K (1600 °C). Thus, according to thermodynamic calculations, some competing reactions are expected during the carbothermic treatment of quartz in a N2-containing atmosphere, which could limit the application of the proposed SiC production method if Si3N4 formation precedes SiC formation.
As is evident from the results presented in Figure 3(a), decreasing d90 of the screened size-fractioned anthracite (decreasing from 682.6 to 119.5 µm) resulted in an increased yield of SiC from 60.3 to 71.2 pct. An associated decrease in Si3N4 formation, i.e., from 34.9 to 22.9 pct, was also observed. The remaining unreacted quartz content was approximately 5.4 ± 0.6 pct in all cases.
Considering the results presented in Figure 3(b), SiC formation was relatively insensitive to the particle size of the screened quartz fraction. Although the linear interpolated lines indicate the increased SiC formation with larger size-fractioned quartz, the percentages of SiC formed were almost identical for the quartz size fractions with d90 of 556.8 and 192.5 µm, i.e., 81.76 and 81.17 pct SiC, respectively. The remaining unreacted quartz content was approximately 3.1 ± 1.2 pct for all cases.
The most likely reasons why the investigated reaction system was sensitive to anthracite (Figure 3(a)) and insensitive to quartz (Figure 3(b)) particle sizes can be deduced from the reaction mechanisms (Reactions [4] and [5]). Under the experimental conditions considered (particle size range, temperature, and holding time), volatilization of SiO2 to SiO (Reaction [4]) was complete, and therefore SiC formation was insensitive to quartz particle size (Figure 3(b)). In contrast, SiC formation increased with the decreasing anthracite particle size (Figure 3(a)), which relates to the volatile SiO having a larger anthracite surface area to react with (Reaction [5]). This deduction was proven, since SiC formation takes place on the surface of the anthracite particles (Figure 6 and associated text). The formation of Si3N4 will be discussed in greater detail later, since it has significant mechanistic implications.
In order to further investigate the effect of particle size, milled mixtures of quartz and anthracite (Table I) were carbothermically treated at 1873.15 K (1600 °C). These results are presented in Figure 4. Milling was kept to a minimum to prevent in-situ SiC formation. Raygan et al.[43] produced a small quantity of SiC from silica sand during 200 hours of low-energy ball milling, whereas Lu et al.[44] completely converted silicon powder to SiC under the condition of 24 hours of high-energy stirred bead milling. However, it is highly unlikely that any significant amount of SiC formed during the less-than 60 seconds’ milling used in this study. XRD analysis (quantitative Rietveld refined, as indicated in Section II) confirmed that either no SiC formed during milling, or that the quantities formed were below the detection limit of the analytical technique.

Percentage compositions (formed SiC and Si3N4, as well as unreacted quartz) of milled mixtures carbothermically treated at 1873.15 K (1600 °C) indicated on the primary y-axis, and particle sizes (d90) of these milled mixtures indicated on the secondary y-axis. The bars indicate the standard deviations from three experimental repeats
As is evident from the results presented in Figure 4, carbothermic treatment at 1873.15 K (1600 °C) of milled quartz and anthracite mixtures with d90 ≤ 350.9 µm resulted in almost complete conversion of quartz to SiC (> 98 pct), with very limited Si3N4 formation (≤ 1.4 pct). The results indicate that limited milling would be required to ensure sufficient interparticle contact to promote the solid-state reaction between SiO2 and C to form SiO, as indicated in Reaction [4]. This result is very promising from a possible industrial application perspective, since limited milling requires less energy input (which is directly related to cost of production). To place these results in perspective, extensive milling to achieve a d90 of 75 µm is specified for the composite pellet mixture used in the industrial solid-state reduction of chromite (also known as chromite prereduction).[18]
Effect of Temperature
To determine the effects of reaction temperature on conversion of quartz, the milled mixture with d90 of 350.9 µm (Table I, 10 seconds’ milling) was carbothermically treated at various temperatures, i.e., 1673.15, 1723.15, 1773.15, and 1873.15 K (1400, 1450, 1500, 1600 °C), as indicated in Figure 5. Previously, it was proven that quartz conversion to SiC at this particle size was almost complete after 1873.15 K (1600 °C) treatments (Figure 4).

Effects of temperature on quartz conversions (average and standard deviation composition, based on three repeats) of the milled mixtures with a d90 of 350.9 µm
As is evident from the temperature-dependency results (Figure 5), Si3N4 was the dominant Si-containing phase after carbothermic treatment at 1673.15 K (1400 °C) (77.1 pct Si3N4 and 4.6 pct SiC), while a significant fraction of unreacted quartz remained (18.3 pct). This indicates that Si3N4 formation occurred at temperatures lower than 1823.15 K (1550 °C), which was predicted from thermodynamic calculations with HSC (Reaction [6]).
It is clear from Figure 5 that for the carbothermic treatment of quartz in a N2 atmosphere, Si3N4 forms before SiC, further emphasizing the importance of understanding Si3N4 formation and its possible conversion to SiC.
Mechanistic Deductions
The mechanism for the reaction system considered in this paper needed to be established, or at least the important aspects identified. To initiate this, backscattered SEM and cross-sectional EDS mappings of carbothermically treated milled mixtures with d90 of 350.9 µm were considered, as presented in Figure 6. Again, the d90 of 350.9 µm was considered due to the high quartz-to-SiC conversion and the minimal milling energy required to achieve this particle size (Figure 4 and associated text).

Backscattered SEM micrograph of SiC-containing particles originating from a milled mixture with d90 of 350.9 µm that was carbothermically treated at 1873.15 K (1600 °C) (a); a polished cross section of a SiC-containing particle (b); and the corresponding EDS mapping for C (c), O (d), and Si (e)
SEM-EDS spot analyses were performed on the surface of a typical unpolished SiC-containing particle (marked with a white outline in Figure 6(a)), as well as areas toward the center of a cross-sectioned (polished) SiC-containing particle (Figure 6(b)). These EDS results are presented in Table II and should be considered as semiquantitative, with the O content determined by difference. The areas selected for investigation are referred to as “Surface” (from unpolished sample as in Figure 6(a)), as well as “Transition” and “Core” (from polished sample as in Figure 6(b)), in Table II.
Considering the carbon-based resin used to prepare samples prior to polishing, the C content of the “Transition” and “Core” sampled areas may be a minor overestimation, and the O continent may also originate at least partially from the resin used. Some Si detected at the core of the investigated particle (Figure 6(b)) might also be a result of smearing (spreading of traces thereof) of elements during polishing. Notwithstanding these data limitations, the compositions of the “Surface” and “Transition” areas confirm the enrichment of Si and C in these areas, which indicates SiC formation (pure SiC contains 70.04 pct Si and 29.96 pct C). The Al content, which is likely to originate from impurities in the anthracite and quartz, also decreased toward the core of the investigated particles.
Considering the results presented in Table II, the decrease in Si content and associated C content increase toward the center of the particles was of particular mechanistic interest. This is indicative of a shrinking core mechanism, where the C is nonmobile and the Si is the mobile element. This supports the commonly held mechanistic theory that the SiO2 present in the quartz was reduced to form SiO(g) in the presence of C(s) at elevated temperatures 1873.15 K (1600 °C), as indicated in Reaction [4].[32,40] The in-situ-generated SiO(g) then subsequently reacted with C(s) present on the anthracite surface to form SiC(s), as indicated in Reaction [5].[32,40] SiO(g) can further penetrate the outer SiC(s) layer on the anthracite particle to react with underlying C(s), until either the SiO(g) reaches its penetration limit, or the supply of SiO(g) is depleted. It is also likely that SiC(s) formation is dependent on the availability of reaction surface on the anthracite, with a smaller particle size implying a larger surface area. However, the effect of particle size (Figures 3 and 4) indicated that after a critical particle size is achieved (which was established as d90 ≤ 350.9 µm), conversion of quartz to SiC was almost complete. Therefore the remaining C “Core” (Figure 5) is due to the excess C supply (Section II–C), indicating that all Si was converted to SiC, before a SiO(g) penetration limit had been achieved.
The abovementioned reaction mechanism usually occurs in a CO/CO2 atmosphere. However, in this study a N2 atmosphere was used (Section II–C). Nevertheless, some CO and CO2 will form in situ due to the C reacting with oxygen in the material matrix. At a pressure of 1 atm and over the temperature range considered in this study [i.e., 1723.15 to 1873.15 K (1450 to 1600 °C)], the equilibrium of the Boudouard reaction (Reaction [7]) is shifted toward the right, implying that almost all CO2 will be converted to CO, which results in a more reducing environment favorable for SiC formation.
Although the discussion of the aforementioned mechanism, within the context of commonly accepted mechanistic theory (Reactions [4] and [5])[32,40] and recognition of some in-situ-formed CO/CO2, is valid, it was proven beyond doubt that Si3N4 was the dominant Si-containing phase at maximum carbothermic reaction temperatures of approximately 1738.15 K (≤ 1465 °C) (Figure 5). It is therefore likely that Si3N4 formation would precede SiC formation, as the milled mixtures were heated from room temperature to 1873.15 K (1600 °C) (Figures 3 and 4). The mechanistic role of Si3N4 within this process therefore needed to be clarified.
Figure 7 presents the surface morphologies of a milled mixture with d90 of 350.9 µm carbothermically treated at 1673.15 and 1723.15 K (1400 to 1450 °C). Thin, strand-like growths or whiskers are clearly visible in the material treated at 1673.15 K (1400 °C) (Figure 7(a)). EDS spot analyses were performed on various areas, indicated by the white numbers 1 to 4 (Figures 7(a) and (b)), as presented in Table III.

SEM micrographs of milled mixture material with d90 of 350.9 µm carbothermically treated at 1673.15 K (1400 °C) (a) and 1723.15 K (1450 °C) (b). The compositions of the numbered areas were determined by EDS analysis, as indicated in Table III
By jointly considering Figure 7(a) and “Spot” analysis 1 in Table III, it is evident that the whiskers that are formed at 1673.15 K (1400 °C) (Figure 7(a), numbered area 1) consist mainly of Si3N4. These Si3N4 whiskers sit on and around unreacted C particles (areas numbered 2 and 4 in Figures 7(a) and (b), as well as Table III). At 1723.15 K (1450 °C) these whiskers lose their whisker-like morphology, as indicated by area number 3 in Figure 7(b) and Table III. This morphology change may be ascribed to the dissociation of nitrogen from the Si3N4 molecule. The observed morphology change for this dissociation was previously described as occurring at temperatures between 1873.15 to 2073.15 K and (1600 to 1800 °C).[45] However, Murray et al.[45] referred to Si3N4 dissociation in a solar-thermal process where Si3N4 was obtained from purer sources of SiO2 and C. Therefore, the lower temperature Si3N4 dissociation observed here may be due to kinetic catalytic effects of impurities present in quartz and anthracite. Notwithstanding the uncertainty regarding the cause, the lower temperature Si3N4 dissociation resulted in a very finely divided source of Si being deposited onto the anthracite particles. Subsequently, this resulted in almost complete conversion of Si to SiC at 1873.15 K (1600 °C), which is below the expected SiC formation temperature. Therefore, Si3N4 formation significantly enhanced SiC formation and should thus not be regarded as a competing, unwanted reaction.
In order to assess whether the formation of Si3N4, its subsequent decomposition, and the SiC formation occurred in distinct phases, TG analysis of milled mixture material with a d90 of 350.9 µm was undertaken, as presented in Figure 8. From the presented data, it is clear that up to approximately 1073.15 K (800 °C), very little mass loss, which is mainly a result of devolatilization of the anthracite, occurs. However, from approximately ≥ 1473.15 K (≥ 1200 °C), significant and continuous (not phased) mass loss occurs. The only deductions that can be made from this TG data, in conjunction with previous results, were that Si3N4 formation initiates at ≥ 1473.15 K (≥ 1200 °C), followed by its decomposition at ≤ 1723.15 K (Figure 7(b) and Table III) and formation of SiC, which is complete at ≥ 1873.15 K (≥ 1600 °C) (Figures 3 and 4). Therefore, the formation of Si3N4, its decomposition, and SiC formation did not occur in distinct separate temperature phases, but rather concurrently.

TG curve of milled mixture material with d90 of 350.9 µm that was carbothermically treated in a N2 atmosphere at increasing temperature, in the temperature range 1073.15 to 1873.15 K (800 to 1600 °C)
Considering all the mechanistic deductions presented thus far, a simplified mechanism was proposed. The first likely step is that SiO2 reacts with N2 to form Si3N4 (Reaction [8]) in the presence of carbon at ≥ 1473.15 K (≥ 1200 °C), as indicated in Figure 7(a). Further increase in temperature results in the dissociation of Si3N4 into Si and N2, which occurs at ≤ 1723.15 K (1450 °C) (Figure 7(b)). However, instead of Si metal remaining after the aforementioned dissociation, SiC forms (Reaction [10]). These three reaction steps, i.e., Reactions [8, 9, and 10], account for the SiC formation at lower than conventional temperature. In the current study, > 30 pct SiC formed at these reduced temperatures, if a milled mixture was considered (Figure 5). Thereafter, as the reaction temperature increases further, the conventional reaction mechanism, represented by Reactions [11 and 12], takes effect. This results in complete conversion of SiO2 to SiC at ≥ 1873.15 K (≥ 1600 °C) (Figure 5).
To further assess whether the alternative reaction mechanism as proposed here, i.e., Reactions [8, 9, and 10], is possible, a thermodynamic equilibration calculation was performed using the thermodynamic software program HSC. For this calculation, it was assumed that 11.86 kmole C reacts with 1 kmole of SiO2 (corresponding to the actual mole ratio used in this study) in the presence of surplus N2 (taken as 100 kmole). As is evident from the graphical representation of the results obtained (Figure 9), the thermodynamic calculation confirms that Si3N4 can form at lower temperatures than SiC and that it will decompose in the temperature range in which SiC starts forming. Although such thermodynamic calculations have some limitations, (e.g., they do not consider kinetic inhibiting or catalytic effects), the results certainly support the mechanism proposed here.

Equilibria according to thermodynamic calculations using the thermodynamic software program HSC
Possible Practical Applications of On-Site SiC Production
This paper has mainly focused on exploring the fundamental possibility of producing SiC from waste materials (i.e., CO-rich off gas, as well as screened out quartz and anthracite fines). However, to guide future studies in this field of research, it is worthwhile considering the limitations of the current study and the aspects that must still be considered, before process upscaling is deliberated.
Possibly the most significant fundamental scientific concern is that all the results presented thus far were obtained in a N2 gaseous environment. Such an environment will not be industrially feasible. When this study was initiated, it was thought that a N2 atmosphere would be disadvantageous due to possible competing Si3N4 formation. However, as was indicated, Si3N4 formation actually enhances SiC formation (Section III–C). This indicates that N2 in the ambient gaseous environment (with approximately 78 pct N2) will not adversely influence SiC formation. The approximately 21 pct oxygen (O2) in the ambient environment could, however, be problematic since oxidation of the carbonaceous source (anthracite in this case) could occur during CO combustion to supply the heat. To test if this will be a problem, milled material mixture with a d90 of 350.9 µm (identified as being optimal, i.e., reducing milling cost, but maintaining SiC formation efficiency) was placed in a crucible with a normal crucible lid on top. The mixture filled approximately 70 pct of the crucible’s total volume, and the fit of the crucible lid was not airtight, allowing air to enter the reaction vessel. After treatment using the same temperature profile applied to all previous samples (up to 1873.15 K, 1600 °C), XRD analysis revealed SiC and Si3N4 contents of 97.7 and 2.3 pct, respectively. No residual SiO2 was detected. The high SiC content and the absence of SiO2 proved that oxidation that would interfere with SiC formation can be prevented. Within the milled material, in-situ-generated CO2 and CO (Reaction [7]) will generate a positive partial pressure, preventing O2 from entering the mixture. In addition, it seems that a similar positive partial pressure within the crucible, combined with the significant excess C (Section II–C), also prevented oxidation on the surface of the material mixture. The proposed process therefore seems feasible if some precautions are taken to prevent oxidation.
The availability of sufficient volumes of CO-rich off-gas, as well as fine quartz and anthracite, may also be considered as limiting factors for possible on-site SiC production. The availability and quality of off-gas is determined by two factors: (i) furnace operating conditions such as power input and (ii) the metallurgical health of the furnace. A major benefit of the proposed approach of storing energy in the form of SiC would be that SiC can be produced when the waste materials (CO-rich off-gas, as well as fine quartz and anthracite) are available, and the process can be halted if they are not.
Selecting a proper furnace design for SiC production would be critical. Although rotary kilns are often used in prereduction processes, a rotary kiln would require pelletized material feed, since unpelletized material tumbling in such a furnace would cause huge dust issues. An alternative approach would be to use a rotary hearth furnace, in which the material is stationary and the refractory-lined bed moves through several refractory-lined chambers. Such a furnace may contain several chambers, each with its own operating temperature, allowing a systematic increase in operational temperature. Figure 10 presents a simple schematic of a rotary hearth furnace and the suggested operational temperatures.

A simplified schematic of a rotary hearth furnace and the suggested operating temperatures for on-site SiC production
It was previously mentioned that precautions should be taken to prevent oxidation on the surface of the material mixtures thermally treated, and that dust suppression should also be considered. Obviously, the material could be pelletized. Alternatively, a sacrificial layer of coarse, low-grade coal could be placed on top of the material in the rotary hearth, as indicated in Figure 11. This material will assist in the formation of a partial positive CO2/CO atmosphere below the mixed material surface.

Schematic illustration of material in rotary hearth furnace for the proposed SiC production route
Many other parameters need to be investigated before the process can be tested on a larger scale. For instance, heat transfer between combusted CO-rich off-gas and the solid reagents (quartz and anthracite in this case study) needs to be considered, with equipment design and residence time optimization required to enable attaining the optimum material temperature, i.e., ≥ 1873.15 K (≥ 1600 °C) according to the findings presented in Section III.
EDS mapping of C and Si (Figure 7) of the formed SiC indicated that C-rich particles are coated with SiC. Therefore, unreacted C particles, which can be separated from the SiC, are unlikely to occur. It is therefore essential that actual reductive smelting of ores be conducted in future to assess the effectiveness of the unique SiC-coated and unreacted C core-type particles. Procedures for tracking possible changing SiC to C ratios in such feed material would also have to be developed, since different ratios are bound to impact the reductant metallurgical balance in the smelting furnace. Obviously, a technoeconomic study should also be undertaken if these investigations yield positive results.
Conclusions
In this paper, we present a novel approach to capture and store the energy associated with CO-rich off-gas combustion in the form of SiC, which can be used as a partial replacement for carbonaceous reductant during the smelting process.
SiC formation was achieved using materials partially classified as waste (quartz and anthracite fines) from a large FeCr producer. Relatively little milling of the raw materials was required (minimizing milling energy) and carbothermic treatment at 1873.15 K (1600 °C) resulted in the almost complete conversion of quartz to SiC. This temperature can be achieved without difficulty by combusting CO-rich off-gas. It was found that Si3N4 will form in a N2-containing atmosphere prior to SiC formation. However, the subsequent decomposition of Si3N4 actually enhanced the SiC formation. Therefore, Si3N4 formation should not be considered as a competing pathway or a hampering product.
A quick assessment of the possible practical application of this process indicated that numerous practical aspects still have to be considered before it can be implemented, but the study presented here indicates considerable potential.
References
S.E. Haggerty: Reviews in Mineralogy and Geochemistry, 1991, vol. 25, pp. 355–416.
J.P. Beukes, N.F. Dawson, and P.G. Van Zyl: J. S. Afr. Inst. Min. Metall., 2010, vol. 110, pp. 743–750.
J.P. Beukes, S.P. du Preez, P.G. van Zyl, D. Paktunc, T. Fabritius, M. Päätalo, and M. Cramer: J. Clean Prod., 2017, vol. 165, pp. 874–889. https://doi.org/10.1016/j.jclepro.2017.07.176.
R.T. Jones: DC Arc Furnaces—Past, Present, and Future. in: Mackey, P.J., E.J. Grimsey, R.T. Jones, and G.A. Brooks eds., Celebrating the Megascale. Springer, USA 2014. http://dx.doi.org/10.1007/978-3-319-48234-7.
J. Basson, and J. Daavittila: in High Carbon Ferrochrome Technology-Chapter 9, M. Gasik ed., Handbook of Ferroalloys: Theory and Technology. Elsivier, United Kingdom. 2013. http://dx.doi.org/10.1016/B978-0-08-097753-9.00009-5.
Y. van Staden, J.P. Beukes, P.G. van Zyl, J.S. du Toit, and N.F. Dawson: Miner. Eng., 2014 vol. 56, pp. 112-120. https://doi.org/10.1016/j.mineng.2013.11.004.
B.B. Lind, A.M Fällman, and L.B. Larsson: Waste Manag., 2001, vol. 21, pp. 255–264. https://doi.org/10.1016/S0956-053X(00)00098-2.
J. Zelić: Cement. Concrete. Res., 2005, vol. 35, pp. 2340–2349. https://doi.org/10.1016/j.cemconres.2004.11.019.
P. Niemelä, and M. Kauppi: Proceedings of the 11th International Ferro Alloys Conference, New Delhi, India, 2007, pp. 171–79.
C. Maine, J. Smit, and E. Giesekke: The Solid Stabilization of Soluble Wastes Generated in the South African Ferrochrome Industry. Water Research Commission, 2005.
11. J.P. Beukes, P.G. Van Zyl, and M. Ras: J. S. Afr. Inst. Min. Metall., 2012, vol. 112, pp. 347–352.
S.P. Du Preez, J.P. Beukes, W.P.J. Van Dalen, P.G. Van Zyl, D. Paktunc, and M.M. Loock-Hatting: Water SA, 2017, vol. 43, pp. 298–309. http://dx.doi.org/10.4314/wsa.v43i2.13.
M. Riekkola-Vanhanen: Finnish expert report on best available techniques in ferrochromium production, Helsinki, 1999.
P. Niemelä, H. Krogerus, and P. Oikarinen: Proceedings of 10th International Ferroalloys Congress, Cape Town, South Africa, February 2004, pp. 68–77.
G. Kapure, C. Kari, S.M. Rao, and K. Raju: Proceedings of the 11th International Ferro Alloys Conference, New Delhi, India, 2007, pp. 165–70.
E. Schubert, and R. Gottschling: in Co-generation: a challenge for furnace off-gas cleaning systems. Southern African Pyrometallurgy, R.T. Jones, and P. den Hoed eds., Southern African Pyrometallurgy. Johannesburg: Southern African Institute of Mining and Metallurgy, 2011, pp. 6–9.
L. Shoko, J.P Beukes, and C.A. Strydom. Miner. Eng., 2013, vol 49, pp. 33–39. https://doi.org/10.1016/j.mineng.2013.04.015.
E.L.J. Kleynhans, J.P. Beukes, P.G. van Zyl, P.H.I. Kestens, and J.M. Langa: Miner. Eng., 2012, vol. 34, pp. 55–62. https://doi.org/10.1016/j.mineng.2012.03.021.
S.P. Du Preez, J.P. Beukes, and P.G. van Zyl: Metall. Mater. Trans. B, 2015, vol. 46, pp. 1002–1010. https://doi.org/10.1007/s11663-014-0244-3.
D. Hall, K. Donaldson, J. McAllister, T. Marrs, J.A. Ross, D. Anderson, D. Baker, L.-C. Chen, P.J. Baxter, and V. Murray: Toxicology, Survival and Health Hazards of Combustion Products. Royal Society of Chemistry, 2015. ISSN: 1757-7179.
O.K. Kordina and S.E. Saddow: Advances in Silicon Carbide Processing and Applications, S.E. Saddow and A. Agarwal, eds., Artech House, London, 2004. ISBN: 1-58053-740-5.
P. Šajgalík, J. Sedláček, Z. Lenčéš, J. Dusza, and H.-T. Lin: J. Eur. Ceram. Soc., 2016, vol. 36, pp. 1333–1341. https://doi.org/10.1016/j.jeurceramsoc.2015.12.013.
O. Demir: Ferroalloy production. Patent application WO2001079572 A1, 2001.
D.M. Pheiffer, and K. Cookson: Ferrochrome Alloy Production. Patent application WO2015159268 A1, 2015.
W. Malan, G. Akdogan, S. Bradshaw, and G. Bezuidenhout: J. S. Afr. Inst. Min. Metall., 2015, vol. 115, pp. 375–383.
R.I. Glastonbury, J.P. Beukes, P.G. van Zyl, L.N. Sadikit, A. Jordaan, Q.P. Campbell, H.M. Stewart, and N.F. Dawson: J. S. Afr. Inst. Min. Metall., 2015, vol. 115, pp. 699–706.
E.L.J. Kleynhans, J.P. Beukes, P.G. van Zyl, J.R. Bunt, N.S.B. Nkosi, and M. Venter: Metall. Mater. Trans. B, 2017, vol. 48, 827–840. https://doi.org/10.1007/s11663-016-0878-4.
X. Li, G. Zhang, K. Tang, O. Ostrovski, and R. Tronstad: Metall. Mater. Trans. B, 2015, vol. 46, pp. 1343–1352. https://doi.org/10.1007/s11663-015-0293-2.
S. Bandyopadhyay, and J. Mukerji: Ceram. Int., 1991, vol. 17, pp. 171–179. https://doi.org/10.1016/0272-8842(91)90064-7.
R.V. Krishnarao, Y.R. Mahajan, and T.J. Kumar: J. Eur. Ceram. Soc., 1998, vol. 18, pp. 147–152. https://doi.org/10.1016/S0955-2219(97)00093-9.
Z. Wu, S. Deng, N. Xu, J. Chen, J. Zhou, and J. Chen: Appl. Phys. Lett., 2002, vol. 80, pp. 3829–3831. http://dx.doi.org/10.1063/1.1476703.
S.H. Chen, and C.I. Lin: J. Mater. Sci Lett., 1997, vol. 16, pp. 702–704. https://doi.org/10.1023/A:1018508525803.
G.S. Gupta, P. Vasanth Kumar, V.R. Rudolph, and M. Gupta: Metall. Mater. Trans. A, 2001, vol. 32, pp. 1301–1308. https://doi.org/10.1007/s11661-001-0220-9.
A. Roine: HSC Chemistry, 7th ed, Outotec Research, Pori, Finland, 2009, pp. 11-1–11-25.
A. Agarwal, and U. Pad: Metall. Mater. Trans. B, 1999, vol. 30(2), pp. 295–306.
W. Biermann, R.D. Cromarty, and N.F. Dawson. J. S. Afr. Inst. Min. Metall., 2012, vol. 112, pp. 301–308.
B.M. Moshtaghioun, R. Poyato, F.L. Cumbrera, S. de Bernardi-Martin, A. Monshi, M.H. Abbasi, F. Karimzadeh, and A. Dominguez-Rodriguez: J. Eur. Ceram. Soc., 2012, vol. 32, pp. 1787–1794. https://doi.org/10.1016/j.jeurceramsoc.2011.12.021.
K. Itaka, T. Ogasawara, A. Boucetta, R. Benioub, M. Sumiya, T. Hashimoto, H. Koinuma, and Y. Furuya: J. Phys. Conf. Ser., 2015, vol. 596, IOP Publishing, 012015. https://doi.org/10.1088/1742-6596/596/1/012015.
R. Hill, and C. Howard: J. Appl. Crystallogr., 1987, vol. 20, pp. 467–474. https://doi.org/10.1107/S0021889887086199.
F. van Dijen, and R. Metselaar: J. Eur. Ceram. Soc., 1991, vol. 7, pp. 177–184. https://doi.org/10.1016/0955-2219(91)90035-X.
T. Muranaka, Y. Kikuchi, T. Yoshizawa, N. Shirakawa, and J. Akimitsu: Sci. Technol. Adv. Mater., 2008, vol. 9, pp. 1–8. https://doi.org/10.1088/1468-6996/9/4/044204.
H. Arik: J. Eur. Ceram. Soc., 2003, vol. 23, pp. 2005–2014. https://doi.org/10.1016/S0955-2219(03)00038-4.
S. Raygan, B. Kondori, and H.M. Yangijeh: Int. J. Refract. Met. Hard Mater., 2012, vol. 29, pp. 10–13. https://doi.org/10.1016/j.ijrmhm.2010.06.002.
Y. Lu, Y. Wang, Z. Pan, H. Shen, and L. Wu: Ceram. Int., 2013, vol. 39, pp. 4421–4426. https://doi.org/10.1016/j.ceramint.2012.11.033.
J.P. Murray, G. Flamant, and C.J. Roos: Solar Energy, 2006, vol. 80, pp. 1349–1354. https://doi.org/10.1016/j.solener.2005.11.009.
Acknowledgments
The authors thank the FeCr producer that provided the materials used in this study. The financial assistance of the National Research Foundation (NRF) toward the Ph.D. study of S.P. du Preez (Grant Number: 101345) is hereby acknowledged. Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the NRF. Funding from the Metal Production Centre for Research-based Innovation (SFI) (237738) of the Norwegian Research Council is acknowledged. The authors would also like to thank Mr. Kosie Oosthuizen from the Geology Subject Group for allowing the use of their pulverizer, and Mr Nico Lemmer from the Faculty of Engineering for allowing the use of their particle size analyzer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted December 7, 2017.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
du Preez, S.P., Beukes, J.P., van Zyl, P.G. et al. Silicon Carbide Formation Enhanced by In-Situ-Formed Silicon Nitride: An Approach to Capture Thermal Energy of CO-Rich Off-Gas Combustion. Metall Mater Trans B 49, 3151–3163 (2018). https://doi.org/10.1007/s11663-018-1413-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-018-1413-6