
Abstract
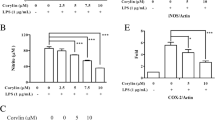
The inflammatory reaction plays an important role in the pathogenesis of the neurodegenerative disorders. tert-butylhydroquinone (tBHQ) exhibits a wide range of pharmacological activities including anti-oxidative and anti-inflammatory action. In this study, we tried to elucidate possible effects of tBHQ on lipopolysaccharide (LPS)-induced inflammatory reaction and its underlying mechanism in neuron-like PC12 cells. tBHQ inhibited LPS-induced generation of reactive oxygen species (ROS) and elevation of intracellular calcium level. It also inhibited LPS-induced cyclooxygenase 2 (COX-2), TNF-α, nuclear factor KappaB (NF-kB), and caspase-3 expression in a dose-dependent manner while stabilizing nuclear factor-erythroid 2 p45-related factor 2. Moreover, the phosphorylations of p38, ERK1/2, and JNK were suppressed by tBHQ. These results suggest that the anti-inflammatory properties of tBHQ might result from inhibition of COX-2 and TNF-α expression, inhibition of NF-kB nuclear translocation along with suppression of MAP kinases (p38, ERK1/2, and JNK) phosphorylation in PC12 cells, so may be a useful agent for prevention of inflammatory diseases.









Similar content being viewed by others

Abbreviations
- Aβ:
-
Amyloid β
- AREs:
-
Antioxidant responsive elements
- CAT:
-
Catalase
- CNS:
-
Central nervous system
- ECL:
-
Electrochemiluminescence
- GSH:
-
Glutathione
- H2O2 :
-
Hydrogen peroxide
- Keap1:
-
Kelch-like ECH-associated protein 1
- MDA:
-
Malondialdehyde
- MTT:
-
3-[4,5-Dimethylthiazol-2-yl]-2, 5-dephenyl tetrazolium bromide
- NF-κB:
-
Nuclear factor-κB
- NGF:
-
Nerve growth factor
- Nrf2:
-
Nuclear factor-erythroid 2 p45-related factor 2
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- tBHQ:
-
tert-Butylhydroquinone
References
Shimohama S (2000) Apoptosis in Alzheimer’s disease—an update. Apoptosis 5(1):9–16
Ashe KH (2001) Learning and memory in transgenic mice modeling Alzheimer’s disease. Learn Mem 8(6):301–308
Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinters HV, Frautschy SA, Cole GM (1998) Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer’s disease. Am J Pathol 152(2):379–389
Irizarry MC, Hyman BT (2001) Alzheimer disease therapeutics. J Neuropathol Exp Neurol 60(10):923–928
Guha M, Mackman N (2001) LPS induction of gene expression in human monocytes. Cell Signal 13(2):85–94
Allan SM, Rothwell NJ (2003) Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci 358(1438):1669–1677
Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN (2008) Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76(11):1485–1489
Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, Jain MR, Liew C, Chan JY, Kong AN (2007) Toxicogenomics of endoplasmic reticulum stress inducer tunicamycin in the small intestine and liver of Nrf2 knockout and C57BL/6 J mice. Toxicol Lett 168(1):21–39
Khodagholi F, Eftekharzadeh B, Maghsoudi N, Rezaei PF (2010) Chitosan prevents oxidative stress-induced amyloid beta formation and cytotoxicity in NT2 neurons: involvement of transcription factors Nrf2 and NF-kappaB. Mol Cell Biochem 337(1–2):39–51
Stewart D, Killeen E, Naquin R, Alam S, Alam J (2003) Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem 278(4):2396–2402
Tusi SK, Ansari N, Amini M, Amirabad AD, Shafiee A, Khodagholi F (2010) Attenuation of NF-κB and activation of Nrf2 signaling by 1,2,4-triazine derivatives, protects neuron-like PC12 cells against apoptosis. Apoptosis 15:738–751
Koh K, Cha Y, Kim S, Kim J (2009) tBHQ inhibits LPS-induced microglial activation via Nrf2-mediated suppression of p38 phosphorylation. Biochem Biophys Res Commun 380(3):449–453
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Kutuk O, Basaga H (2003) Aspirin prevents apoptosis and NF-kappaB activation induced by H2O2 in HeLa cells. Free Radic Res 37:1267–1276
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol 186:421–431
Kakkar P, Das B, Viswanathan PN (1984) A modified spectrophotometric assay of superoxide dismutase. Ind J Biochem Biophys 21:130–132
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Guroff G (1985) PC12 cells as a model of neuronal differentiation. In: Bottenstein JE (ed) Cell culture in neurosciences. Plenum Press, New York, pp 245–272
Martin TF, Grishanin RN (2003) PC12 cells as a model for studies of regulated secretion in neuronal and endocrine cells. Methods Cell Biol 71:267–286
Omata Y, Saito Y, Fujita K, Ogawa Y, Nishio K, Yoshida Y, Niki E (2008) Induction of adaptive response and enhancement of PC12 cell tolerance by lipopolysaccharide primarily through the upregulation of glutathione S-transferase A3 via Nrf2 activation. Free Radic Biol Med 45:1437–1445
Miller DK (1997) The role of caspase family of cysteine proteases in apoptosis. Semin Immunol 9:35–49
Yu LC, Turner JR, Buret AG (2006) LPS/CD14 activation triggers SGLT-1-mediated glucose uptake and cell rescue in intestinal epithelial cells via early apoptotic signals upstream of caspase-3, Exp. Cell Res 312:3276–3286
Eftekharzadeh B, Maghsoudi N, Khodagholi F (2010) Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2 N neurons. Biochimie 92(3):245–253
Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J (2005) Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci 83(2):313–328
Liu CS, Chen NH, Zhang JT (2007) Protection of PC12 cells from hydrogen peroxide-induced cytotoxicity by salvianolic acid B, a new compound isolated from Radix Salviae miltiorrhizae. Phytomedicine 14(7–8):492–497
Blum D, Torch S, Nissou MF, Verna JM (2001) 6-Hydroxydopamine-induced nuclear factor-kappa B activation in PC12 cells. Biochem Pharmacol 62(4):473–481
Cárcamo JM, Pedraza A, Bórquez-Ojeda O, Zhang B, Sanchez R, Golde DW (2004) Vitamin C is a kinase inhibitor: dehydroascorbic acid inhibits IkappaBalpha kinase beta. Mol Cell Biol 24(15):6645–6652
Shishodia S, Potdar P, Gairola CG, Aggarwal BB (2003) Curcumin (diferuloylmethane) down-regulates cigarette smoke-induced NF-kappaB activation through inhibition of IkappaBalpha kinase in human lung epithelial cells: correlation with suppression of COX-2, MMP-9 and cyclin D1. Carcinogenesis 24(7):1269–1279
Li Y, Liu L, Barger SW, Mrak RE, Griffin WS (2001) Vitamin E suppression of microglial activation is neuroprotective. J Neurosci Res 66(2):163–170
Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M, Quattrone A, Calabrese V (2006) Curcumin activates defensive genes and protects neurons against oxidative stress. Antioxid Redox Signal 8:395–403
Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73(7):2424–2428
Cowley S, Paterson H, Kemp P, Marshall CJ (1994) Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77(6):841–852
Heneka MT, Löschmann PA, Gleichmann M, Weller M, Schulz JB, Wüllner U, Klockgether T (1998) Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem 71(1):88–94
Männel DN, Rosenstreich DL, Mergenhagen SE (1979) Mechanism of lipopolysaccharide-induced tumor necrosis: requirement for lipopolysaccharide-sensitive lymphoreticular cells. Infect Immun 24(2):573–576
Kobayashi M, Yamamoto M (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzym Regul 46:113–140
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284(20):13291–13295
He X, Lin GX, Chen MG, Zhang JX, Ma Q (2007) Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol Sci 98(1):298–309
Ho HK, White CC, Fernandez C, Fausto N, Kavanagh TJ, Nelson SD, Bruschi SA (2005) Nrf2 activation involves an oxidative-stress independent pathway in tetrafluoroethylcysteine-induced cytotoxicity. Toxicol Sci 86(2):354–364
Jiang T, Huang Z, Chan JY, Zhang DD (2009) Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol Appl Pharmacol 240(1):8–14
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120(5):649–661
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441(7092):431–436
Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, Dean K, Franzoso G (2006) The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ 13(5):712–729
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G (2004) Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119(4):529–542
Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H (2003) NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 22(15):3898–3909
Claudio E, Brown K, Siebenlist U (2006) NF-kappaB guides the survival and differentiation of developing lymphocytes. Cell Death Differ 13(5):697–701
Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18(18):2195–2224
Kucharczak J, Simmons MJ, Fan Y, Gélinas C (2003) To be, or not to be: NF-kappaB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene 22(56):8961–8982
Gilmore TD (2006) Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25(51):6680–6684
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22(2):153–183
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103(2):239–252
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81(2):807–869
Ramachandiran S, Huang Q, Dong J, Lau SS, Monks TJ (2002) Mitogen-activated protein kinases contribute to reactive oxygen species-induced cell death in renal proximal tubule epithelial cells. Chem Res Toxicol 15(12):1635–1642
Zanotto-Filho A, Delgado-Cañedo A, Schröder R, Becker M, Klamt F, Moreira JC (2010) The pharmacological NFkappaB inhibitors BAY117082 and MG132 induce cell arrest and apoptosis in leukemia cells through ROS-mitochondria pathway activation. Cancer Lett 288(2):192–203
Aggarwal BB, Natarajan K (1996) Tumor necrosis factors: developments during the last decade. Eur Cytokine Netw 7(2):93–124
Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104(4):487–501
Wang H, Joseph JA (2000) Mechanisms of hydrogen peroxide-induced calcium dysregulation in PC12 cells. Free Radic Biol Med 28(8):1222–1231
Tan S, Sagara Y, Liu Y, Maher P, Schubert D (1998) The regulation of reactive oxygen species production during programmed cell death. J Cell Biol 141(6):1423–1432
Castillo MR, Babson JR (1998) Ca(2+)-dependent mechanisms of cell injury in cultured cortical neurons. Neuroscience 86(4):1133–1144
Acknowledgments
This work was supported by Shahid Beheshti University of Medical Sciences Research Funds. The authors thank MS. Joodi for her help in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khodagholi, F., Tusi, S.K. Stabilization of Nrf2 by tBHQ prevents LPS-induced apoptosis in differentiated PC12 cells. Mol Cell Biochem 354, 97–112 (2011). https://doi.org/10.1007/s11010-011-0809-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-011-0809-2