
Abstract
Purpose
Dysfunctional mitochondria are considered to be the major source of intracellular reactive oxygen species and play a central role in the pathophysiology of myocardial ischemia/reperfusion. This study sought to determine effects of mitochondria-targeted cytoprotective peptide SBT-20 on myocardial infarct size in two different models of ischemia/reperfusion.
Methods
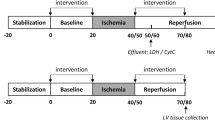
For in vivo studies, anesthetized Sprague Dawley rats were subjected to 30 min of coronary artery occlusion followed by 3 h of reperfusion. Rats received saline (control), low dose SBT-20 (0.3 mg/kg/h) or high dose SBT-20 (3 mg/kg/h) treatment (n = 15 rats in each group). Saline or SBT-20 were delivered into the jugular vein starting 5 min after coronary artery occlusion and were continued for one hour post coronary artery reperfusion. Body temperature, heart rate and blood pressure were monitored during the procedure. At the end of 3 h reperfusion, the ischemic risk area, no-reflow area, and infarct size were measured. In separate in vitro studies, isolated rat hearts were exposed to 20 min global ischemia, followed by SBT-20 administration (1 μM) or no SBT-20 (control) throughout the 2 h reperfusion. In vitro studies were conducted in cells and heart mitochondria to ascertain the mitochondrial effects of SBT-20 on mitochondrial respiration and reactive oxygen species production.
Results
In the in vivo study, the ischemic risk areas (as a percentage of the left ventricle) were similar among the saline (49.5 ± 2.3 %), low dose SBT-20 (48.6 ± 2.1 %), and high dose SBT-20 groups (48.7 ± 3.0 %). Treatment with SBT-20 significantly reduced infarct size ( as a percentage of risk area) in low dose (62.1 ± 4.4 %) and high dose (64.0 ± 4.9 %) compared with saline treatment (77.6 ± 2.6 %, p = 0.001 for both doses). There was no difference in infarct size between low and high dose SBT-20 treatment. The no-reflow areas (as a percentage of the risk area) were comparable among the saline (23.9 ± 1.7 %), low dose SBT-20 (23.7 ± 2.8 %), and high dose groups (25.0 ± 2.1 %). Body temperature, heart rate and blood pressure were comparable among the 3 groups at baseline, during ischemia, and at the end of 3 h of reperfusion. In the in vitro study, infarct size was reduced from 43.3 ± 2.6 % in control group (n = 11) to 17.2 ± 2.8 % in the SBT-20 treatment group (n = 5, p < 0.05). There were no benefits of SBT-20 on recovery of left ventricular developed pressure, coronary flow, or maximal rates of contraction/relaxation. In cell studies, treatment with SBT-20 significantly improved maximal mitochondrial respiration in response to an H2O2 challenge. In isolated mitochondria, reactive oxygen species production was significantly blunted following treatment with SBT-20.
Conclusions
In summary, SBT-20 significantly reduced infarct size in two different models of myocardial injury, but did not affect hemodynamics or no-reflow area in rat heart. The reduction in injury is postulated to involve stabilization of mitochondrial function and reduced mitochondrial production of ROS.



Similar content being viewed by others


References
Kloner RA, Dai W, Hale SL, Shi J. Approaches to improving cardiac structure and function during and after an acute myocardial infarction: acute and chronic phases. J Cardiovasc Pharmacol Ther. 2016;21(4):363–7.
Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the "dark side" of reperfusion. Circulation. 2009;120(21):2105–12.
Goldhaber JI, Weis JN. Oxygen free radicals and cardiac reperfusion abnormalities. Hypertension. 1992;20:118–27.
Petrosillo G, Venosa ND, Pistolese M, et al. Protective effect of melatonin against mitochondrial dysfunction associated with cardiac ischemia–reperfusion: role of cardiolipin. FASEB J. 2006;20:269–76.
Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10(3):601–19.
Cho J, Won K, Wu D, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18(3):215–20.
Frasier CR, Moukdar F, Patel HD, et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res. 2013;98(1):47–55.
Brown DA, Hale SL, Baines CP, et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther. 2014;19(1):121–32.
Kloner RA, Hale SL, Dai W, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J Am Heart Assoc. 2012;1(3):e001644.
Sloan RC, Moukdar F, Frasier CR, et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52(5):1009–18.
Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297–312.
Alleman RJ, Tsang AM, Ryan TE, et al. Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol. 2016;310(10):H1360–70.
Krumschnabel G, Fontana-Ayoub M, Sumbalova Z, et al. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol Biol. 2015;1264:245–61.
Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–5.
Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96(6):672–83.
Brown DA, Sabbah HN, Shaikh SR. Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol Ther. 2013;140(3):258–66.
Zhao K et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279(33):34682–90.
Zhao K, Luo G, Zhao GM, Schiller PW, Szeto HHJ. Pharmacol Exp Ther. 2003;304:425–32.
Birk AV et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24(8):1250–61.
Birk AV, Chao WM, Liu S, Soong Y, Szeto HH. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim Biophys Acta. 2015;1847(10):1075–84.
Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. J Pharmacol Exp Ther. 2009;329(1):94–101.
Kloner RA. No-reflow phenomenon: maintaining vascular integrity. J Cardiovasc Pharmacol Ther. 2011;16(3–4):244–50.
Gibson CM, Giugliano RP, Kloner RA, et al. EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J. 2016;37(16):1296–303.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
These studies were supported by grants from NIH (R01 HL123647 and R15 HL122922 to David A. Brown) and Stealth BioTherapeutics (to David A. Brown, and Robert A. Kloner).
Conflict of Interest
Authors David A. Brown and Robert A. Kloner have received research grants from Company Stealth BioTherapeutics, and have received consulting income from Stealth BioTherapeutics. Authors Wangde Dai, Elissa Cheung, Justin B. Perry, Mitchell E. Allen, Rick J. Alleman declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Dai, W., Cheung, E., Alleman, R.J. et al. Cardioprotective Effects of Mitochondria-Targeted Peptide SBT-20 in two Different Models of Rat Ischemia/Reperfusion. Cardiovasc Drugs Ther 30, 559–566 (2016). https://doi.org/10.1007/s10557-016-6695-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-016-6695-9