
Abstract
Colletotrichum fungi belonging to the Colletotrichum gloeosporioides species complex include a number of economically important postharvest pathogens that often cause anthracnose. Until now, different species within this group could only be distinguished from one another reliably using multigenic phylogenetic analyses. Using a comparative genomics approach, we developed a marker that can differentiate Colletotrichum fructicola, Colletotrichum aenigma and Colletotrichum siamense within the C. gloeosporioides species complex based on PCR amplicon size differences. When we used this marker to classify 115 isolates collected over 20 years from strawberries in Chiba Prefecture, Japan, the isolates were predominantly C. fructicola. To our knowledge, this is the first report characterizing different species of Colletotrichum infecting strawberries in Japan and contributes to our understanding on the diversity of anthracnose pathogens in Japan.
Similar content being viewed by others


Introduction
Colletotrichum is a genus of fungi that often cause anthracnose disease. In Japan and elsewhere, strawberry anthracnose has been attributed to infections by Colletotrichum gloeosporioides, causing at least one major epidemic of anthracnose crown rot in the United States during the 1970s (Smith 2008; Suzuki et al. 2010). Anthracnose disease is a major problem in the Japanese strawberry cultivation industry where infections are thought to be spread primarily through latently infected plants and infected plant material (Suzuki et al. 2010). The presence of even one infected plant can result in the infection of an entire field, highlighting the importance of early detection and classification.
Phylogenetic analyses using molecular markers have revealed that many members of the genus Colletotrichum may be grouped into independent lineages with similar evolutionary histories (Cai et al. 2011). Several of these lineages have now been systematically classified as species complexes comprising closely related sibling species (Weir et al. 2012). Many species within the C. gloeosporioides species complex have previously been classified as C. gloeosporioides sensu lato, largely based on morphological characteristics and known host range (Hyde et al. 2009). In addition, the study by Weir et al. (2012) showed that ITS sequences alone, which have long been used to classify fungal species, are not very reliable in distinguishing different species within this species complex. However, it is important to distinguish between species within the same species complex since different species may warrant different infection management strategies (Cai et al. 2011). For example, it is known that, while Colletotrichum kahawae is an aggressive pathogen of coffee, Colletotrichum asianum, Colletotrichum fructicola and Colletotrichum siamense are opportunistic pathogens and thus do not require the same level of control when encountered in the field (Prihastuti et al. 2009).
In Japan, strawberry anthracnose has been attributed to C. gloeosporioides and, at least to our knowledge, there have not been studies to determine whether different members of the C. gloeosporioides species complex are associated with strawberry infections. In previous studies, however, rep-PCR analysis indicated that even within Japan alone, infections are caused by strains that can be subdivided into groups that can be distinguished genetically (Suzuki et al. 2010, 2012). In this study, we found that at least three distinct species, C. fructicola, C. siamense and Colletotrichum aenigma, were responsible for strawberry anthracnose disease in the Chiba Prefecture in Japan. Using genomic analyses, we designed a set of PCR markers that can distinguish these species within the C. gloeosporioides species complex. Using these markers, we identified isolates that were collected in Chiba Prefecture, Japan, between 1994 and 2014. Our data suggest that C. fructicola has been the predominant species in this region during the last two decades.
Materials and methods
Isolation of anthracnose pathogens from infected strawberry
Here we use “isolate” to refer to a pure culture of a single sample from an infected plant; the term “strain” refers to a defined isolate that can be distinguished from other isolates using phylogenetic methods, in this case, based on six combined loci (ACT, CAL, CHS-1, ITS, TUB and GAPDH). Isolates from the C. gloeosporioides species complex were collected from anthracnose lesions on strawberry (Fragaria × ananassa) or randomly sampled, latently infected strawberry plants from the Inba, Chiba, Kimitsu, Awa, Chosei, Sanbu and Kaiso regions in Chiba Prefecture between 1994 and 2014. From latently infected plants, conidia were isolated using the ethanol immersion technique (Ishikawa 2003). These isolates were identified as members of the C. gloeosporioides species complex using the primer CgInt and the universal primer ITS4 (Mills et al. 1992).
Comparative genomic analysis
Pairwise alignments of genome assemblies were carried out using the nucmer program with default settings (Kurtz et al. 2004). Conserved regions present in the genomes of C. fructicola (Cg38), C. aenigma (Cg56; MAFF 305913) and C. siamense (Cg363; P. Gan et al. unpublished data) were identified using the bedtools suite of programs (Quinlan and Hall 2010). The sequences encompassing the marker regions have been deposited in GenBank (accessions KX247845–KX247850). Nucleotide BLAST searches were carried out using the nucleotide sequences between the predicted primer binding sites as queries against specific individual genome assemblies in CLCGenomicsWorkbench8 (QIAgen) (E-value cutoff = 1E-5).
Phylogenetic analysis
Sequences from actin (ACT), chitin synthase (CHS-1), calmodulin (CAL), glyceraldehyde-3-phosphate (GAPDH), β-tubulin 2 (TUB) and the internal transcribed spacer (ITS) were amplified from isolates using primers in Table 1 from fungal DNA extracted using the DNeasy kit (QIAgen). PCR reactions were set up with 2× PCR mix (Promega), 0.5 µM each of forward and reverse primers (Table 1) and 25–75 ng of purified fungal DNA per tube. DNA sequences from these isolates have been deposited in GenBank (accessions KU642469–KU642552 and KX513878–KX513929). PCR was carried out at 95 °C for 3 min; followed by 35 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min; and a final extension step at 72 °C for 5 min. The DNA sequences obtained were aligned with sequences of previously classified isolates (Weir et al. 2012) using the program MAFFT version 7.215 (Katoh et al. 2002) and trimmed using the trimAl program version 1.2rev59 (Capella-Gutiérrez et al. 2009) with the automated1 settings. Concatenating the sequences resulted in an alignment consisting of 2854 positions, which were then partitioned according to genes and used for phylogenetic tree analysis using a maximum likelihood analysis in RAxML version 8.2.4 (Stamatakis 2014) and a Bayesian inference analysis using MrBayes version 3.2.6 (Ronquist and Huelsenbeck 2003). The program Partitionfinder version 1.1.1 (Lanfear et al. 2012) was used to identify the best nucleotide substitution models for the analysis of each partition using the BIC criteria (Lanfear et al. 2012). Both RAxML and MrBayes analyses were performed using the GTRGAMMA + I model for each partition, allowing each partition to evolve under different rates. MrBayes was run twice for 20,000,000 generations with samples taken every 1000 generations with the first 25% of generations discarded as burnin. Trees were visualized in FigTree version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Sequences from Colletotrichum boninsense (Weir et al. 2012) were included as an outgroup.
Genotyping of isolates
PCR reactions were carried out using extracted fungal DNA and the Marker 1 and Marker 2 primer pairs (Table 1) for 30 cycles at 95 °C for 1 min, 55 °C for 30 s and 72 °C for 30 s; followed by a final extension step at 72 °C for 5 min before separation in a 1% agarose gel. Specific primers for virulent strains (Suzuki et al. 2008) were used to distinguish between virulent and less virulent isolates.
Results
Marker region identification
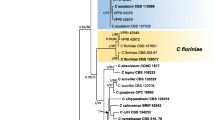
The genomes of three strains isolated from infected strawberry plants, Cg38 (isolated from Chiba), Cg56 (isolated from Tochigi) and Cg363 (isolated from Chiba), which belonged to different groups based on a previous rep-PCR analysis (Suzuki et al. 2012), were sequenced. These strains were then classified using the multigenic phylogenetic analysis of the ACT, CAL, CHS-1, ITS, GAPDH and TUB sequences (Fig. 1). In the multigenic phylogenetic trees (Figs. 1, 2), the three strains were separated into three separate species within the C. gloeosporioides species complex: C. fructicola (Cg38), C. aenigma (Cg56) and C. siamense (Cg363).

Bayesian inference phylogenetic tree showing the relationship between selected strains from the Colletotrichum gloeosporioides species complex based on actin, chitin synthase 1, calmodulin, GAPDH and ITS sequences. Numbers at each node indicates the Bayesian posterior probability values expressed in percentages. Scale bar indicates number of expected changes per site

Maximum likelihood phylogenetic tree generated with RAxML software showing the relationship between selected strains from the Colletotrichum gloeosporioides species complex. The tree was drawn based on actin, chitin synthase 1, calmodulin, GADPH, β-tubulin 2 and ITS sequences. Values on branches are bootstrap support values expressed in percentages (1000 bootstrap replicates). Scale bar indicates number of expected changes per site
The Cg38 strain was found to be closely related to the previously sequenced C. fructicola Nara gc5 (Gan et al. 2013) and was used as a reference strain in subsequent analyses. Pairwise alignments of the genome assemblies were carried out using the C. fructicola (Cg38) assembly as a reference strain. A total of 5987 genomic regions identified using the nucmer analysis (Fig. 3), comprising 6.35 Mb of sequence that were present in the reference strain, were not conserved in the C. siamense and C. aenigma assemblies. Among the 5897 genomic regions, 2203 genomic regions were specific to the reference C. fructicola strain. Of the other 3784 regions, 3110 unconserved regions were flanked by at least 2 kb of sequence in the reference strain. Of these 3110 unconserved regions, 1025 target regions were between 200 bp and 1 kb in size. As a condition for primer design, it was necessary to identify target regions flanked by highly conserved regions. Thus, sequences including these 1025 target regions were queried for overlap with 6153 genomic regions that were conserved among the three strains mentioned. A total of 1365 potential conserved regions were found to flank the target regions (Fig. 3).

Strategy to identify regions for marker candidate regions
Among these, two loci were manually selected for further analysis. Approximately 1.5–4 kb of the selected loci from the reference strain were then used as queries for a BLAST search of the available genome assemblies. Primers were then designed based on sequences that were conserved in all isolates and present in single-copy regions. Primers were tested on a number of isolates that were classified based on phylogenetic trees based on ACT, CAL, CHS-1, ITS, TUB and GADPH sequences including sequences from members of the previously classified C. gloeosporioides species complex (Weir et al. 2012).
Test of markers
Marker 1
Test of markers
In the C. fructicola genome, the region targeted by Marker 1 was found to overlap with the CGGC5_05943 gene, which encodes a hypothetical protein. The protein encoded by CGGC5_05943 is conserved in the genomes of strains outside of the C. gloeosporioides species complex, such as C. graminicola, C. higginsianum, C. orbiculare, C. sublineola and C. fioriniae. However, there is sufficient divergence at the nucleotide level such that nucleotide BLAST results of the region amplified by Marker 1 against that of other Colletotrichum genome sequences including C. higginsianum, C. graminicola (O’Connell et al. 2012), C. incanum (MAFF 238712; Gan et al. 2016), C. orbiculare, C. fructicola Nara gc5 (Gan et al. 2013), C. sublineola (Baroncelli et al. 2014a), C. fioriniae (Baroncelli et al. 2014b), and C. gloeosporioides (Alkan et al. 2013) showed that the amplified region was specific to C. fructicola, C. aenigma and C. siamense. Indeed, according to BLAST analysis (cutoff E-value = 1E-5), the sequence was also absent in the genome assembly of the closely related C. gloeosporioides strain sequenced by Alkan et al. (2013). In PCR reactions, Marker 1 amplified a single band from various C. gloeosporioides species complex members including C. kahawae strains (Fig. 4a).

Amplification of bands from DNA of fungal isolates from the Colletotrichum gloeosporioides species complex using Marker 1 (a), which amplifies bands from all C. gloeosporioides strains tested, and Marker 2 (b), which can distinguish between C. fructicola, C. siamense and C. aenigma based on amplicon size. c Actin sequences were amplified as a control
Marker 2
PCR with Marker 2 showed amplification of bands of ~1 kb from Cg38 (C. fructicola), ~550 bp in Cg363 (C. siamense) and ~300 bp from Cg56 (C. aenigma; Fig. 3) strains. The locus containing Marker 2 is located in an intergenic region and includes a region with multiple related sequences in other parts of the genome assemblies according to the BLAST search, indicating that while the primer binding regions are specific, they flank a potential repeat region. To confirm that the different PCR amplicon sizes are correlated with different species, at least five isolates from each pattern (Table 2) were classified using multilocus phylogenetic trees that were drawn using Bayesian and maximum likelihood methods (Figs. 1, 2). Both methods were found to result in trees that were consistent in terms of their topologies (Figs. 1, 2). Cg192, Cg231, Cg255, Cg131 and NK24, which all showed the same sized amplicon of ~550 bp (Fig. 4b), were all classified as C. siamense according to the multilocus phylogenetic trees (Figs. 1, 2). In addition, amplicons of ~300 bp were amplified from the DNA of Cg111, Cg112, Cg132, Cg191 and NK18, which all clustered together with known strains that have previously been identified as C. aenigma. All four of the isolates that did not show amplification with Marker 2 but that could be amplified by Marker 1 were classified as C. kahawae (Fig. 4a, b). The lack of PCR products indicate that the region amplified by Marker 2 may be specific to members within the Musae subclade in the C. gloeosporioides species complex (Figs. 1, 2).
Genotyping of isolates from the Chiba Prefecture between 1994 and 2014
A total of 115 Colletotrichum isolates from strawberry plants in Chiba Prefecture were identified as belonging to the C. gloeosporioides species complex using the CgInt and ITS4 marker sequences (Mills et al. 1992). These were then classified as virulent or less virulent using a previously designed primer pair (Suzuki et al. 2008). According to these results (Fig. 5), 88 isolates were identified as virulent and 27 as less virulent isolates. When the isolates were then tested using Marker 2, 81 of the 88 virulent isolates tested (92%) were classified as C. fructicola, while the remaining seven were classified as C. siamense based on the amplified PCR band sizes (Fig. 5a). On the other hand, 14 of the 27 less virulent isolates were identified as C. aenigma, while eight were identified as C. siamense and one as C. fructicola (Fig. 5b). The identification of C. fructicola as the predominant virulent species of anthracnose growing on strawberry in the Chiba Prefecture was consistent from 1994 to 2014.

Numbers and types of virulent (a) and less virulent (b) isolates of Colletotrichum from the field by year
Discussion
In recent years, the need to reclassify plant pathogens such as Colletotrichum using molecular-based techniques has been recognized because previous classifications based on morphology and host range have been found to be likely incorrect or less definitive (Ko Ko et al. 2011). Studies to characterize which specific species are associated with infections are important since different species may be associated with different infection outcomes in the field as mentioned for coffee anthracnose (Prihastuti et al. 2009).
To divide strains into different groups, other studies have made use of random amplified polymorphic DNA (RAPD) polymorphisms, internal transcribed spacer-restriction fragment length (ITS-RFLP) polymorphisms or classification by ITS sequences (Gautam 2014; Smith 2008). While RAPD and ITS-RFLP polymorphisms give an indication of genetic variability among isolates, specific polymorphisms have not yet been associated with specific species. Further, the classification of strains using a single marker sequence has been found to be insufficient to distinguish strains belonging to different species within the C. gloeosporioides species complex (Weir et al. 2012).
Using a comparative genomics strategy, we successfully developed a single marker to distinguish among three common pathogens of strawberry, C. fructicola, C. siamense and C. aenigma. The described marker flanks a short repeat that is expanded in C. fructicola relative to C. siamense and C. aenigma. It is likely that this sequence was gained/expanded in this group because it was absent in closely related C. kahawae strains. It is believed that the comparative genomics method utilized in this study is a practical strategy that can be further utilized to develop markers for other distinguishing traits or species differences.
The multilocus phylogenetic analysis of 20 isolates isolated from Japan indicated that these isolates tended to group with one another within each species, hinting at single, rather than multiple introductions of the different species into the field. This result is perhaps unsurprising given that the isolates were from a restricted geographical region, but remains to be confirmed with further phylogenetic analyses since this analysis was restricted to only 20 isolates.
From the analysis of isolates collected within Chiba Prefecture over 20 years, we showed that at least four species within the C. gloeosporioides species complex can infect strawberry plants in the field. This mirrors findings on mango anthracnose, which had previously been attributed to C. gloeosporioides infections (Lima et al. 2013). In more recent years, mango anthracnose has been revealed to be caused by different species which also belong to the C. gloeosporioides species complex, namely C. asianum, C. fructicola, C. tropicale, C. karstii and C. dianesei (Lima et al. 2013). Different species also seem to contribute to strawberry anthracnose infections. Recent studies have found that different species of the C. gloeosporioides species complex are associated with strawberry plants including C. fructicola, C. siamense and C. theobromicola in the United States (Weir et al. 2012), C. fructicola in Korea (Nam et al. 2013) and C. murrayae, C. gloeosporioides, C. fructicola and C. aenigma in China (Han et al. 2016). In our study, we found that species from both the Musae and to a lesser extent, the Kahawae clades can infect strawberries in Japan. In addition, although C. fructicola was identified as the predominant virulent species in the field, both virulent and less virulent isolates of C. fructicola and C. siamense were identified in the present study, indicating that specific species are not strictly correlated with virulence on strawberry plants, unlike what has been observed for coffee anthracnose (Prihastuti et al. 2009). The identification of multiple species of Colletotrichum infecting strawberry plants indicates the diversity of strawberry anthracnose pathogens, which in turn suggests that this population of pathogens can adapt to changing conditions (McDonald and Linde 2002), which may then inform disease management strategies.
References
Alkan N, Meng X, Friedlander G, Reuveni E, Serenella S, Sherman A, Thon M, Fluhr R, Prusky D (2013) Global aspects of pacC regulation of pathogenicity genes in Colletotrichum gloeosporioides as revealed by transcriptome analysis. Mol Plant-Microbe Interact 26:1345–1358
Baroncelli R, Sanz-Martín JM, Rech GE, Serenella SA, Thon M (2014a) Draft genome sequence of Colletotrichum sublineola, a destructive pathogen of cultivated sorghum. Genome Announc 2:e00540–14
Baroncelli R, Sreenivasaprasad S, Sukno SA, Thon MR, Holub E (2014b) Draft genome sequence of Colletotrichum acutatum sensu lato (Colletotrichum fioriniae). Genome Announc 2:e00112–14
Cai L, Giraud T, Zhang N, Begerow D, Cai G, Shivas RG (2011) The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers 50:121–133
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Carbone I, Kohn LM (1999) A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91:553–556
Gan P, Ikeda K, Irieda H, Narusaka M, O’Connell RJ, Narusaka Y, Takano Y, Shirasu K (2013) Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol 197:1236–1249
Gan P, Narusaka M, Kumakura N, Tsushima A, Takano Y, Narusaka Y, Shirasu K (2016) Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome Biol Evol 8(5):1467–1481
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118
Gautam AK (2014) Colletotrichum gloeosporioides: biology, pathogenicity and management in India. J Plant Physiol Pathol 2:2
Glass NL, Donaldson GC (1995) Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microbiol 61:1323–1330
Han YC, Zeng XG, Xiang FY, Ren L, Chen FY, Gu YC (2016) Distribution and characteristics of Colletotrichum spp. associated with anthracnose of strawberry in Hubei. China Plant Dis 100:996–1006
Hyde K, Cai L, Cannon P, Crouch JA, Crous PW, Damm U, Goodwin PH, Chen H, Johnston PR, Jones EBG, Liu ZY, McKenzie EHC, Moriwaki J, Norieung P, Pennycook SR, Pfenning LH, Prihastuti H, Sato T, Shivas RG, Tan YP, Taylor PWJ, Weir BS, Yang YL, Zhang JZ (2009) Colletotrichum—names in current use. Fungal Divers 39:147–182
Ishikawa S (2003) Method to diagnose latent infection by Glomerella cingulata in strawberry plants using ethanol. J Gen Plant Pathol 69:372–377
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Ko Ko TW, Stephenson SL, Bahkali AH, Hyde KD (2011) From morphology to molecular biology: can we use sequence data to identify fungal endophytes? Fungal Divers 50:113–120
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL (2004) Versatile and open software for comparing large genomes. Genome Biol 5:R12
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Lima NB, Batista MV, De Morais MA et al (2013) Five Colletotrichum species are responsible for mango anthracnose in northeastern Brazil. Fungal Divers 61:75–88
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40:349–379
Mills PR, Sreenivasaprasad S, Brown AE (1992) Detection and differentiation of Colletotrichum gloeosporioides isolates using PCR. FEMS Microbiol Lett 98:137–143
Nam MH, Park MS, Lee HD, Yu SH (2013) Taxonomic re-evaluation of Colletotrichum gloeosporioides isolated from strawberry in Korea. Plant Pathol J 29:317–322
O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Tores MF, Damm U, Buiate EA, Epstein L, Alkan N, Altmüller J, Alvarado-Balderrama L, Bauser CA, Becker C, Birren BW, Chen Z, Choi J, Crouch JA, Duvick JP, Farman MA, Gan P, Heiman D, Henrissat B, Howard RJ, Kabbage M, Koch C, Kracher B, Kubo Y, Law AD, Lebrun M-H, Lee Y-H, Miyara I, Moore N, Neumann U, Nordström K, Panaccione DG, Panstruga R, Place M, Procter RH, Prusky D, Rech G, Reinhardt R, Rollins JA, Rounsley S, Schardl CL, Schwartz DC, Shenoy N, Shirasu K, Sikhakolli UR, Stüber K, Sukno SA, Sweigard JA, Takano Y, Takahara H, Trail F, Does HC, Voll LM, Will I, Young S, Zeng Q, Zhan J, Zhou S, Dickman MB, Schulze-Lefert P, Themaat EVL, Ma LJ, Vaillancourt LJ (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet 44:1060–1065
O'Donnell K, Cigelnik E (1997) Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol Phylogenet Evol 7:103–116
Prihastuti H, Cai L, Chen H, McKenzie EHC, Hyde KD (2009) Characterization of Colletotrichum species associated with coffee berries in northern Thailand. Fungal Divers 39:89
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics Oxf Engl 19:1572–1574
Smith BJ (2008) Epidemiology and pathology of strawberry anthracnose: a North American perspective. HortScience 43:69–73
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Suzuki T, Tanaka-Miwa T, Ito Y, Uematsu S, Hirayama Y, Okayama K (2008) Development of specific PCR primers designed for the detection of Colletotrichum gloeosporioides causing strawberry anthracnose (abstract in Japanese). Jpn J Phytopathol 74:198
Suzuki T, Tanaka-Miwa C, Ebihara Y, Ito Y, Uematsu S (2010) Genetic polymorphism and virulence of Colletotrichum gloeosporioides isolated from strawberry (Fragaria × ananassa Duchesne). J Gen Plant Pathol 76:247–253
Suzuki T, Kanegae Y, Yoshida N, Suzuki T, Ohtani T (2012) Phylogenetic analysis of partial sequences of elongation factor 1 α and pathogenicity of the Colletotrichum gloeosporioides isolated from strawberry plants (abstract in Japanese). Jpn J Phytopathol 78:184
Templeton MD, Rikkerink EHA, Solon SL, Crowhurst RN (1992) Cloning and molecular characterization of the glyceraldehyde-3-phosphate dehydrogenase-encoding gene and cDNA from the plant pathogenic fungus Glomerella cingulata. Gene 122:225–230
Weir BS, Johnston PR, Damm U (2012) The Colletotrichum gloeosporioides species complex. Stud Mycol 73:115–180
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: A Guide to Methods and Applications. Academic Press, New York, pp 315–322
Acknowledgements
This work was supported in part by Council for Science, Technology and Innovation (CSTI), Cross-ministerial Strategic Innovation Promotion Program (SIP), “Technologies for creating next-generation agriculture, forestry and fisheries” (Funding agency: Bio-oriented Technology Research Advancement Institution, NARO), the Science and Technology Research Promotion Program for the Agriculture, Forestry, Fisheries, and Food industry to N.N and K.S, and Grant-in-Aid for Scientific Research (KAKENHI) (24228008 and 15H05959 to K.S).
Author information
Authors and Affiliations
Corresponding author
Additional information
The nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession numbers KU642469–KU642552, KX247845–KX247850 and KX513878–KX513929.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gan, P., Nakata, N., Suzuki, T. et al. Markers to differentiate species of anthracnose fungi identify Colletotrichum fructicola as the predominant virulent species in strawberry plants in Chiba Prefecture of Japan. J Gen Plant Pathol 83, 14–22 (2017). https://doi.org/10.1007/s10327-016-0689-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-016-0689-0