
Abstract
A nanocrystalline TiO2 (anatase) nanosheet exposing mainly the (001) crystal faces was tested as photoanode material in dye-sensitized solar cells. The nanosheets were prepared by hydrothermal growth in HF medium. Good-quality thin films were deposited on F-doped SnO2 support from the TiO2 suspension in ethanolic or aqueous media. The anatase (001) face adsorbs a smaller amount of the used dye sensitizer (C101) per unit area than the (101) face which was tested as a reference. The corresponding solar cell with sensitized (001)-nanosheet photoanode exhibits a larger open-circuit voltage than the reference cell with (101)-terminated anatase nanocrystals. The voltage enhancement is attributed to the negative shift of flatband potential for the (001) face. This conclusion rationalizes earlier works on similar systems, and it indicates that careful control of experimental conditions is needed to extract the effect of band energetic on the current/voltage characteristics of dye-sensitized solar cell.
Similar content being viewed by others


Introduction
Dye-sensitized solar cell (DSC), also called Grätzel cell, represents an attractive alternative of solid state photovoltaics due to high efficiency, low cost, and easy fabrication [1–4]. Optimization of TiO2 photoanode for dye-sensitized solar cell has been a subject of numerous studies, and nanocrystalline anatase turned out to be the best material for this device [5]. The voltage enhancement is crucial for further improvement of DSCs [1, 3, 6–10]. Promising data were acquired recently by replacing the traditional electrolyte redox relay, I −3 /I- by other systems [3, 6–10], but little is known about the voltage enhancement via tuning of the photoanode material.
The (101) face is dominating in ordinary TiO2 (anatase) materials (>94 % of the total surface area of crystals) [11]. The remaining face on the anatase crystal is (001) which is consistent with the conclusion that a truncated bipyramid is the corresponding crystal morphology [12]. Only rarely, the rhombic-shaped crystals are found, exposing the (010) face [13]. Both (001) and (010) facets are called “high-energy” or “reactive” ones, and they show interesting activity in catalysis and photocatalysis [13–16].
Comparative studies of anatase (101) or (001) faces have been carried out on macroscopic single-crystal electrodes [17, 18], but the dye sensitization was attempted only on the (101) face of the single-crystal electrode [17]. The works on single-crystal electrodes confirmed that the (001) face had more negative flatband potential and was more active for Li insertion than the (101) face [18]. The conclusion about flatband potential shift was later reproduced on polycrystalline electrodes [19]. Also, the improved Li insertion was subsequently confirmed on polycrystalline (001)-oriented nanosheets [20]. They were prepared hydrothermally in HF medium according to Yang et al. [11], and the follow-up studies [14, 15, 21–23] report on materials enriched up to 90 % with the (001) face.
Recently, Wang et al. [24] studied the sensitization of such (001) nanosheets by CdS quantum dots, but no comparison with (101)-exposing crystals was presented. This was carried out earlier by Yu et al. [25], who observed improved conversion efficiency of a DSC employing hydrothermally grown nanosheets sensitized by the organometallic dye, N719 [chemical name: di-tetrabutylammonium cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′ dicarboxylato) ruthenium(II)], as the photoanode material. They attributed the improvement to three effects specific for the (001)-oriented nanosheets: (1) enhanced diffusion of the electrolyte, (2) more easy adsorption of the dye, and (3) fewer defects in the surface. However, there were no clear experimental data about electrolyte diffusion and defects, and the amount of adsorbed dye (N719) was actually smaller on nanosheets than that on nanoparticles [25] which seems to contradict the statement (2). Yang et al. [23] reported recently on hierarchical spheres with over 90 % (001) faces which were more active than the Degussa P25 titania material in dye-sensitized solar cells, and the effect was ascribed to (1) stronger ability to dissociatively “absorb” (COOH) groups, (2) higher surface area for dye loading, (3) light scattering, and (4) smaller electron losses at grain interfaces [23]. However, these conclusions were not supported by direct experimental data. Zhang et al. [26] tested the (001)-exposed microspheres in a bilayer photoanode with Degussa P25 underlayer, and concluded that the beneficial effect consists in enhanced light scattering on the mirror-like (001) facets.
To address these divergences, we report here on a comparative study showing that a significant effect favoring the (001) nanosheets is the enhanced open-circuit voltage of the solar cell. This result is consistent with the negative shift of flatband potential of the (001) face. To the best of our knowledge, this conclusion is presented here for the first time; hence, it upgrades earlier works dealing with these problems [23, 25, 26].
Experimental section
Materials
Anatase TiO2 nanosheets were prepared as follows: 10 mL of titanium (IV) butoxide was mixed with 0.8–1.2 mL of hydrofluoric acid (concentration, ≈50 %). The mixture was sealed in a Teflon cell encased in a stainless steel autoclave and heated at 180–200 °C for 24 h. A sample was then collected, washed with copious amounts of Milli-Q water and finally dried at 100 °C. The reference material was prepared in the same way, but in the absence of HF which was replaced by the same amount of water. X-ray diffraction patterns (data not shown) confirmed that the reference material was phase-pure anatase. For comparison, a second reference material (coded C240) [27] was also used. It is nanocrystalline anatase, S BET = 89 m2/g, prepared by hydrolysis of titanium tetra(isopropoxide) and hydrothermal recrystallization at 240 °C in the absence of HF [28]. The crystal morphology of C240 is characterized by particles ca. 10–20 nm in size exposing mainly the (101) facets, and it is frequently used for DSC [29]. The photoelectrochemical behavior of both reference materials was very similar.
Electrodes from all parent powder materials (see above) were fabricated using either aqueous paste (A films) or simply ethanolic suspension (E films). The aqueous paste was prepared as follows: 0.3 g of the powder was mixed under slow addition of 4 × 0.15 mL of 10 % aqueous solution of acetylacetone, 0.3 mL of 4 % aqueous solution of hydroxypropylcellulose (MW 100,000), and 0.3 mL of 10 % aqueous solution of Triton-X100. The obtained slurry was deposited on F-doped SnO2 (FTO) glass (TEC 15 from Libbey-Owens-Ford, 15 Ω/sq) with Kapton foil tape defining the TiO2 film edges. The film was then calcined for 30 min in air at 450 °C. For the preparation of E film electrodes, the powder samples were sonicated in ethanol. The obtained slurry was deposited by doctor blading, and a uniform semitransparent film was obtained after drying at room temperature and calcination in air at 450 °C. Typical film thickness was 2–4 μm (Alpha-step profilometer, Tencor Instruments) for both A/E films.
The C101 dye [NaRu(4,4′-bis(5-hexylthiophene-2-yl)-2,2′-bipyridine)(4-carboxylic acid-4′-carboxylate-2,2′-bipyridine)(NCS)2] with chenodeoxycholic acid (cheno) as a coadsorbent was used for sensitization (see [30] for details). Immediately after calcination, the still warm electrode (ca. 50 °C) was dipped in a solution containing 300 μmol/L of C101 dye and 300 μmol/L cheno in a mixed solvent of acetonitrile + t-butanol solution (1:1, v/v). The electrodes were soaked at room temperature in the solution overnight to secure complete attachment of the sensitizer dye to the electrode surface. To avoid irregularities in dye attachment [31], the sensitization time and temperature were identical for all the tested electrodes. The DSC was assembled with platinized F-doped tin oxide (FTO) counter electrode [32, 33] using a Surlyn tape (25 μm in thickness) as a seal and spacer. The electrolyte solution was 0.6 M N-methyl-N-butyl imidazolium iodide, 40 mM I2, 0.075 M lithium iodide, 0.26 M tert-butylpyridine, and 0.05 M guanidine thiocyanate in acetonitrile/valeronitrile (85/15 %, v/v). The cell active area for illumination was 0.22 cm2, defined by a mask.
Methods
The X-ray diffraction (XRD) was investigated on powder samples by XRD-6000, Shimadzu, Japan, using Ni-filtered CuKα radiation (λ = 0.15418 nm). The transmission electron microscopy (TEM) images were measured on a JEOL JEM-2010F microscope. The BET surface areas of the prepared powder materials were determined from nitrogen adsorption isotherms at 77 K using the Micromeritics ASAP 2020 instrument. The surface areas of thin films were determined from Kr-adsorption isotherms, which were measured directly on the sintered films following the methodology described in [34]. The surface concentration of the C101 dye on TiO2 was measured spectrophotometrically as follows: The sensitized electrode was dipped in 0.1 M tetrabutylammonium hydroxide in dimethylformamide and stirred until complete desorption into the liquid took place. The solution was analyzed on the PerkinElmer Lambda 1050 spectrometer using the extinction coefficient ε 550 = 17.5·103 M−1 cm−1 [30]. The dye surface coverage (Г dye) was determined by normalizing the found dye concentration to the film's physical surface area from Kr-adsorption isotherm. Scanning electron microscopy (SEM) and energy-dispersive X-ray analysis (EDX) were carried out with the Hitachi FE SEM S-4800 microscope equipped with the Noran EDX system. Electrochemical measurements were carried out in a one-compartment cell using an Autolab Pgstat-30 (Ecochemie) controlled by GPES-4 software. For photoelectrochemical tests, the light source was a 450-W xenon light source (Osram XBO 450, Germany) with a filter (Schott 113). The light power was regulated to the AM 1.5G solar standard by using a reference Si photodiode equipped with a color-matched filter (KG-3, Schott) to reduce the mismatch in the region of 350–750 nm between the simulated light and AM 1.5G to less than 4 %. The differing intensities were regulated with a neutral wire mesh attenuator. The applied potential and cell current were measured using a Keithley model 2400 digital source meter. Optical determination of the flatband potential was made by a spectroelectrochemical method described in [35–37]. The measurement was carried out in 0.2 M NaClO4 (pH adjusted by HClO4 or NaOH) using UV–vis–NIR spectrometer PerkinElmer Lambda 1050 interfaced to a potentiostat. Measurements were performed in transmission mode at gradually decreasing potentials from 0 V to −1.2 or −1.4 V. Potential was set, and after 1.5 min at the given potential, the spectrum was measured. For data processing, the measured spectra were normalized by subtracting the spectrum measured at 0 V. Nanosecond flash photolysis was applied to C101 dye-sensitized transparent TiO2 layers covered by a film of pure methoxypropionitrile and a thin cover glass. Pulsed excitation (λ = 530 nm, 5-ns FWHM pulse duration, 20-Hz repetition rate) was provided by an optical parametric oscillator (GWU OPO-355), pumped by a frequency-tripled, Q-switched Nd:YAG laser (Continuum, Powerlite 7030). The laser beam output was expanded by plano-concave lens to irradiate a large cross section of the sample, whose surface was kept at a 30° angle to the excitation beam. The probe light, produced by a Xe arc lamp, passed through the first monochromator, various optics, and the sample. Transmitted light was focused on the entrance slit of the second monochromator and detected by a fast photomultiplier tube. Data waves were recorded on a DSA 602A digital signal analyzer (Tektronix). Satisfactory signal-to-noise ratios were typically obtained by averaging over 3,000 laser shots.
Results and discussion
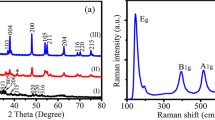
Figure 1a shows a typical X-ray diffraction pattern of TiO2 nanosheets (grown in HF medium), indicating the formation of pure anatase TiO2 (JCPDS no. 21–1272). The diffractograms of (001) nanosheets are similar to those of (101)-nanoparticles, which is in accord with earlier literature [11, 14, 15, 21, 22]. As XRD is obviously not very sensitive to distinguish the (001) or (101) oriented anatase crystals, the morphology of our TiO2 anatase nanosheets was characterized by transmission electron microscopy. A typical nanosheet dimension was 40·30·7 nm3 (Fig. 1b). Figure 1c shows a high-magnification TEM image of two selected individual TiO2 nanosheets (ca. 30·20·7 nm3 in size). The lattice spacing parallel to the top and bottom facets was determined to be ∼0.235 nm, corresponding to the (001) planes of anatase TiO2. Figure 1d shows the corresponding selected-area electron diffraction (SAED) pattern (indexed as the (001) zone axis diffraction). It further indicates that the top and bottom facets of the nanosheets are the (001) planes. The anatase nanosheets exhibited a specific surface area from nitrogen adsorption measurement, S BET between 85 and 120 m2/g depending on the sample batch. Higher values were observed in products grown from mixtures containing less HF and autoclaved at lower temperatures (see “Experimental section”). The typical nanosheet dimension of 40·30·7 nm3 (cf. Fig. 1b) translates into the calculated S BET of 103 m2/g, which matches reasonably well the experimental values from adsorption isotherms. The particles grown in HF-free medium had S BET = 150 m2/g and exhibited the usual bipyramidal crystal shape with (101) facets. This morphology resembles that of C240 particles optimized earlier for DSC applications [29].

Structural characteristics of (001) nanosheets. a XRD pattern. b Low-magnification TEM image. c High-magnification TEM image. d Typical SAED pattern of an individual TiO2 nanosheet
SEM images of our nanosheet-based electrodes are shown in Fig. 2. Both variants of TiO2 film deposition (A films, E films; see “Experimental section”) exhibited a similar morphology of unorganized nanoplatelets which were reasonably uniform and non-agglomerated. This is beneficial for achieving good optical and mechanical quality of the films. It should be noted that even the E films were optically semitransparent with good adhesion to the FTO support in spite of the very simple deposition procedure used and the fact that particles were not stabilized against agglomeration by any other additives as in the case of A films (see “Experimental section”). The as-received nanosheet material contained 8 wt% of F, as determined by EDX analysis, but the heat treatment during electrode fabrication (450 °C, 30 min; see “Experimental section) caused the F content to drop practically to zero for both the A and E films.

Scanning electron microscopy images of the FTO-deposited film from (001)-oriented nanosheets. Left: A film, right: E film
The actual surface area of electrodes (determined from the Kr-adsorption isotherm of the thin-film samples) was normalized to the projected geometric area, which provided the roughness factor. The found values of roughness factor were from 100 to 600 for various film thicknesses, but without significant distinction between the A and E films. More importantly, the surface concentration of the dye (Г dye) was, for both film types (A, E), significantly smaller on the (001) films compared to that on (101) films (Table 1). The found Г dye for (101) films is comparable to the values reported earlier for various Ru-bipyridine sensitizers adsorbed on titania (from 0.56 to 1.16 molecules/nm2; see [38] for the data overview). Recently, Sauvage et al. [31] studied the C101 dye adsorption on a TiO2 film which was, obviously, rich in the (101)-terminated anatase particles. They found an almost perfect dye monolayer (assuming Г dye = 0.57 molecules/nm2) only for the films sensitized at low temperature (4 °C), but the dye coverage exceeded the monolayer saturation limit for the films sensitized at room or higher temperatures. In accord with this report, both our (101) films adsorb more C101 dye than expected for a monolayer (Table 1). However, our Г dye values for the (001) films are below this limit. The relatively smaller dye loading on the (001) face was also reported for the N719 dye [25], but the reason for smaller adsorption capacity of this face is unclear. The anchoring of N719 and N3 dyes [N3 is cis-bis(isothiocyanato) bis(2,2′-bipyridyl-4,4′-dicarboxylato ruthenium(II)] on the (101) face was investigated carefully, including quantum chemical simulations [38, 39], but there is no corresponding study of the (001) face nor that of other dyes like C101.
The trend that smaller values of Г dye are observed for the (001) films qualitatively agrees with the conclusion of Yu at al. [25] who reported that the surface concentration of N719 dye was 143 nmol/cm2 for the (001) nanosheets or 214 nmol/cm2 for the reference (101) nanoparticles, respectively. However, the values reported by Yu et al. [25] do not seem to be normalized to the physical (BET) area of the TiO2 film as in our case. (Note that the recalculated Г dye for the physical surface area would be unrealistically high in the cited work [25], ca. a thousand molecules per square nanometer). Also, we should note that our data in Table 1 do not support the assumption of Yang et al. [23] that the (001) face has better ability to adsorb the N719 dye.
Figure 3 shows the current–voltage characteristics of a solar cell employing the dye-sensitized A films (Fig. 3a) and E films (Fig. 3b). In both cases, data are plotted for (001)-oriented nanosheets (full curves) and reference (101) particles (dashed curves). For the reference (101) particles, we report on the materials grown from Ti(IV) butoxide in HF-free medium (S BET = 150 m2/g), but the plots for our second (101) reference (C240, S BET = 89 m2/g) were quite similar (data not shown). Detailed results about our solar cells are collected in Table 1. It is obvious that the (001)-oriented nanosheet films exhibit smaller short-circuit photocurrents (I sc), particularly at 100 % sun illumination, when the light harvesting is less efficient, and also, the Г dye concentrations are lower (Table 1). Hence, we attribute the smaller I sc to smaller dye loading on the (001) nanosheets. At 10 % sun, the differences in photocurrents are less pronounced or even negligible (A film, Fig. 3a). The (101) particles in A film further show superlinear response to light intensity which might be caused by some dye aggregation. This effect, albeit less pronounced, is also expressed for the remaining films in Table 1. It is further illustrated by higher η values for higher light intensity. Usually, we observe opposite trends [30], but there are also examples when η and light intensity do not scale monotonically for dyes of this type [40]. Furthermore, there is no proportionality between the efficiency, η, and Г dye (Table 1). This finding is supported by Sauvage et al. [31] who reported on decreasing efficiency for the C101-sensitized films, if the Г dye was larger than the monolayer coverage. They concluded that subtle structural characteristics at the C101/TiO2 interface influence the electron transfer dynamics and light harvesting of the assembly [31].

Current–voltage characteristics of dye-sensitized solar cells employing C101-sensitized TiO2 photoanode either from (001)-oriented nanosheets (solid red lines) or reference (101) nanoparticles (dashed blue lines). a A films, b E films
The actually measured photocurrent is further dependent on the TiO2 film thickness. This is difficult to control accurately in various film-deposition techniques; hence, the matching of photocurrents in Fig. 3a (10 % sun) is casual only. From this point of view, we should consider with care the conclusion by Yu at al. [25] who observed a larger photocurrent for the (001) nanosheets, compared to that for (101) nanoparticles and reference commercial titania (P25, Degussa). They reported a film thickness of “about 10 μm,” but the surface concentration of the used dye (N719) was actually smaller by a factor of ca. 1.5 on the (001) nanosheets compared to that on their reference (101) nanoparticles [25]. The relatively smaller dye loading for (001) nanosheets compared to that on (101) particles was also reproduced by us (see Г dye values in Table 1). At full sun illumination, our solar conversion efficiencies η are slightly better for the (101)-terminated nanocrystals. This is due to the dominating contribution of enhanced photocurrent for this face. Our conclusion is in conflict with Yu et al. [25] who reported just the opposite trend in photocurrents and η values.
Figure 3 and Table 1 demonstrate that the most pronounced effect distinguishing (001) nanosheets from the ordinary (101) nanoparticles is the open-circuit voltage (U oc) enhancement of the former. The found differences in open-circuit voltage between nanosheets and nanoparticles (ΔU oc) were 54 or 48 mV (for A films at 10 or 100 % sun, respectively) and 42 or 44 mV for E films at the corresponding conditions and for the actual experiments demonstrated in Table 1. It should be noted that these numbers (and the data in Fig. 3 and Table 1) represent only a single set of measurements; hence, experimental errors and the likely sample-to-sample variations could be unnoticed. To avoid such mistakes, we have carried out a statistical evaluation of data using an array of parallel tests on various TiO2 films with a different preparation history. This analysis showed statistically insignificant differences between A and E films. The averaged values for both film types (A, E) were as follows:
Obviously, even the dependence on the light intensity is not very pronounced too. This points at the hypothesis, that ΔU oc reflects a fundamental physical effect (albeit small) but not a variable introduced by experimental conditions, such as film thickness, preparation history, dye loading, light intensity, etc. More specifically, one of the reasons for positive values of ΔU oc can be the corresponding shift of flatband potential of (001) faces/(101) faces.
Similarly enhanced voltage (ΔU oc = 20 mV) was found for 001-terminated anatase in hierarchical spheres, referred to the value for P25 particles [23]. However, the enhancement was not discussed in [23] and we should also note that P25 is hardly the optimum reference material. It is, actually, a mixture of anatase and rutile, while both phases exhibit a significantly different electronic structure: e.g., the flatband potential of rutile is by 0.2 V more positive than the value for anatase [17].
In contrast to these data, Yu et al. [25] reported on negligible differences in U oc for their (001) nanosheets, (101) nanoparticles, and the P25 film sensitized by N719. To address this paradox, we should note that Yu et al. [25] found a considerably smaller U oc (ca. 580 to 590 mV at 100 % sun) for their DSCs compared to our values (Table 1). Smaller U oc values may stem from a voltage drop caused by enhanced recombination of photoinjected electrons in the titania conduction band with the electrolyte [4, 41], and this effect can mask any other mechanism of fine tuning of U oc, e.g., by crystal face orientation.
Our combination of cheno-coadsorbent with high-extinction dye (C101) provides, obviously, a more-defined interface for exploring small U oc shifts undisturbed by such parasitic effects. Cheno improves the dye attachment by assembling the geometry of the surface complex, prevents agglomeration of dye molecules, and decreases the recombination losses with the electrolyte solution [30, 42, 43]. Furthermore, the back electron transfer can be hindered by a compact titania underlayer on top of the FTO support [4]. This strategy is unsuitable for our study, aiming at the distinction of (001)- and (101)-terminated nanocrystals, but we should note that the optimization of the morphology of TiO2 photoanode may further enhance U oc for a DSC device quite similar to ours [30]. Hence, the elimination of all the other factors influencing ΔU oc is essential, if subtle effects caused by band energetic are to be unraveled.
To test our hypothesis about different band energetic, Fig. 4 compares spectroelectrochemical data for thin-film electrodes made from (001)- and (101)-terminated nanocrystals. This technique was developed for determination of the flatband potential of transparent semiconductors, and it is particularly useful for nanocrystalline electrodes where the standard method based on electrochemical impedance (Mott–Schottky plots) is complicated by various factors [35–37]. The flatband potential of anatase, E fb (in volts) is known to exhibit Nernstian dependence on pH:
where the constant E 0 was reported to be −0.4 V for the (101) face assuming a saturated calomel electrode (SCE) as the reference electrode [17, 35, 36]. The negative shift of absorbance/potential profiles for the (001)-oriented film is apparent from Fig. 4, although the absorbance onset potential is not that clearly different. This might be due to surface states which contribute differently in (001) or (101) films. Based on optical spectra and X-ray photoelectron spectroscopy (XPS), Pan et al. [16] concluded that the conduction band edge of (001) face is upshifted (in electrochemical scale) by 0.04 V. This contradicts our conclusion, as well as earlier spectroelectrochemical reports on single-crystal [18] and polycrystalline [19] electrodes. (One of the reasons for this discrepancy might be that the reported upshift is within experimental error of the determination of the valence band edge by XPS [16].)

Optical absorbance of TiO2 films from (001)- or (101)-oriented nanosheets plotted by open red points or full blue points, respectively. The absorbance was measured at 640 nm and normalized to the mass of the TiO2 film. It is plotted as a function of the applied electrochemical potential in 0.2 M NaClO4 at two different pH values. The lines are guides to the eye
Our data are in qualitative accord with the earlier conclusion by Kawakita et al. [19] based on Mott–Schottky plots for (001)- vs. (101)-oriented nanocrystals similar to ours. These authors reported on negative shift of E fb for their (001)-oriented nanocrystals; the E fb values were from ca. −0.4 to 0 V vs. SCE for various samples at pH 6 (phosphate buffer). This is more positive than the value of E fb ≈ −0.75 V predicted by Eq. (1), but the difference was not commented on in [19]. As discussed above, accurate impedance data are available for single-crystal electrodes only; in this case, the E fb values for (001) face were negatively shifted by 0.06 V compared to that of (101) face [18]. This negative shift of E fb is not far from the observed enhancement of open-circuit voltage, ΔU oc of ca. 0.04 V found in this study (vide ultra). However, there might be also other effects at play, such as different geometry of the dye/titania surface complex [44].
To get further insight into the specific behavior of (001) nanosheets referenced to that of (101) nanoparticles, we employed transient absorption spectroscopy. Nanosecond flash excitation was applied to C101-sensitized TiO2 film contacting pure methoxypropionitrile solvent. Monitoring the transient absorbance signal at a wavelength λ = 650 nm allowed to follow the time course of the dye cation S+ produced upon ultrafast electron injection from the photoexcited sensitizer, S*, into the conduction band of TiO2 (Eq. 2). In the absence of electrolyte (redox mediator), the decay of the S+ signal was due to the back electron transfer reaction (Eq. 3).
The excitation pulse fluence was progressively decreased by neutral density filters until the signal displayed a single exponential decay (Fig. 5). The used fluence of 35 μJ/cm2 was then believed to relate to the injection of at most one electron per TiO2 nanoparticle or nanosheet. The transport of charges between individual nanoparticles or nanosheets is negligible within the sub-millisecond time frame of the back electron transfer process.

Temporal behavior of the transient absorbance measured at λ = 650 nm for a C101 dye-sensitized anatase (001)-nanosheet film, covered by pure methoxypropionitrile solvent, upon ns-pulsed laser excitation (red trace). A typical transient absorbance decay curve obtained in identical conditions with (101) nanoparticles is shown for comparison (blue trace). Amplitudes of both signals were normalized at time zero for a better visual comparison. The first-order kinetic fits are indicated on top of each experimental curve
The corresponding rate constant, k b (Eq. 3), can be obtained from first-order kinetic fit of the absorbance change, ∆A:
where t is time and C and B are constants. The fitted values of k b are: 1.2·103 s−1 for the (001) nanosheets and 7.7·103 s−1 for the (101) nanoparticles, respectively. Hence, the back electron transfer is by a factor of 6 slower for the (001) nanosheets compared to the same process on (101) nanoparticles. The difference in the back electron transfer kinetics observed for both samples could be rationalized by a change in the dye-anchoring geometry. Assuming a damping coefficient β = 1.2 Å–1 for through-space electron tunneling (Eq. 5), retardation of the charge recombination reaction by a factor of 6 corresponds to the increase of the electron transfer distance existing between the Ru(III) center of the oxidized dye and the closest Ti(IV) site on the surface of the oxide by ∆r = 1.5 Å:
C101 has only two carboxylic anchoring groups available on a single bpy ligand. The distance between the Ru ion and the TiO2 thus depends strongly upon the tilt angle of the pyridyl rings on the surface. Because the dicarboxylated bpy ligand (bi-isonicotinic acid) tends to distort upon anchoring on the TiO2 surface, different geometries are expected on the (101) and (001) surfaces [45]. The distance between the Ru center of the dye and the Ti4+ ion directly linked to one of the oxygen atoms of the carboxylic anchor is ≈10 Å when the bpy adopts a flat structure. A change of the tilt angle of the pyridyl rings related to the surface normal of ca. 15° on the (001) facet to ca. 35° on the (101) facet would yield a decrease of the distance between the Ru center in the dye to the surface:
which is the ∆r value calculated from our experimental data (Eq. 5).
In DSCs, the back electron transfer process competes kinetically with the regeneration of the dye by the donor species of the electrolyte. As the latter reaction should not depend upon the crystalline face on which the dye is adsorbed, the retardation of the back electron transfer can only improve the charge separation yield. The difference in back electron transfer rate (k b) hardly influences the open-circuit voltage of the cell. The U OC certainly depends on the injected electrons' quasi-Fermi level [2, 4]. However, the electrons' lifetime is controlled by the competition between their transport in the TiO2 network and their recombination with the oxidized mediator (I –3 ). The recombination between conduction-band electrons and the dye cations, S+, is generally sufficiently intercepted by the reduction of S+ by I− and is not expected to influence the potential of DSC.
Conclusion
Nanocrystalline TiO2 (anatase) in two different crystal morphologies exposing mainly the (001) or (101) crystal faces was employed as a photoanode material in dye-sensitized solar cells. The (001) face adsorbs a smaller amount of the used dye sensitizer (C101) but provides a larger open-circuit voltage of the solar cell. The negative shift of flatband potential is suggested to be responsible for the observed enhancement of U oc. This conclusion helps to rationalize contradictory data in the earlier literature. Furthermore, it indicates that careful control of experimental conditions is needed to extract the effect of band energetic on the current/voltage characteristics of the dye-sensitized solar cell.
References
Grätzel M (2001) Nature 414:338–344
Hagfeldt A, Boschloo G, Sun L, Kloo L, Pettersson H (2010) Chem Rev 110:6595–6663
Yella A, Lee HW, Tsao HN, Yi C, Chandiran AK, Nazeeruddin MK, Diau EWG, Yeh CY, Zakeeruddin SM, Grätzel M (2011) Science 334:629–634
Kalyanasundaram K (2010) Dye sensitized solar cells. EPFL Press & CRC Press, Lausanne
Kavan L (2012) Chem Rec 12:131–142
Wang M, Chamberland N, Breau L, Moser J, Humphry-Baker R, Marsan B, Zakeeruddin SM, Grätzel M (2010) Nature Chem 2:385–389
Daeneke T, Kwon TH, Holmes AB, Duffy NW, Bach U, Spiccia L (2011) Nature Chem 3:211–215
Feldt SM, Gibson EA, Gabrielsson E, Sun L, Boschloo G, Hagfeldt A (2010) J Am Chem Soc 132:16714–16724
Kavan L, Yum JH, Grätzel M (2011) Nano Lett 11:5501–5506
Kavan L, Yum JH, Nazeeruddin MK, Grätzel M (2011) ACS Nano 5:9171–9178
Yang HG, Sun CH, Qiao SZ, Zou J, Liu G, Smith SC, Cheng HM, Lu GQ (2008) Nature 453:638–642
Olson CL, Nelson J, Islam MS (2006) J Phys Chem B 110:9995–10001
Wu B, Guo C, Zheng N, Xie Z, Stucky GD (2008) J Am Chem Soc 130:17563–17567
Han X, Kuang Q, Jin M, Xie Z, Zheng L (2009) J Am Chem Soc 131:3152–3153
Zhang D, Li G, Yang X, Yu JC (2009) Chem Commun 2009:4381–4383
Pan J, Liu G, Lu GQ, Cheng HM (2011) Angew Chem Int Ed 50:2133–2137
Kavan L, Grätzel M, Gilbert SE, Klemenz C, Scheel HJ (1996) J Am Chem Soc 118:6716–6723
Hengerer R, Kavan L, Krtil P, Grätzel M (2000) J Electrochem Soc 147:1467–1472
Kawakita M, Kawakita J, Sakka Y, Shinohara T (2010) J Electrochem Soc 157:H65–H68
Bousa M, Laskova B, Zukalova M, Prochazka J, Chou A, Kavan L (2010) J Electrochem Soc 157:A1108–A1112
Chen JS, Lou XW (2009) Electrochem Commun 11:2332–2335
Yang HG, Liu G, Qiao SZ, Sun CH, Jin YG, Smith SC, Zou J, Cheng HM, Lu GQ (2009) J Am Chem Soc 131:4078–4083
Yang W, Li J, Wang Y, Zhu F, Shi W, Wan F, Xu D (2011) Chem Commun 47:1809–1811
Wang X, Liu G, Wang L, Pan J, Lu GQ, Cheng HM (2011) J Mater Chem 21:869–873
Yu J, Fan J, Lv K (2010) Nanoscale 2:2144–2149
Zhang H, Han Y, Liu X, Yu H, Zhang S, Yao X, Zhao Z (2010) Chem Commun 46:8395–8397
Kavan L, Grätzel M, Rathousky J, Zukal A (1996) J Electrochem Soc 143:394–400
Barbe C, Arendse F, Comte P, Jirousek M, Lenzmann F, Shklover V, Grätzel M (1997) J Amer Ceram Soc 80:3157–3171
Grätzel M (2006) Prog Photovolt Res Appl 14:429–442
Gao F, Wang Y, Shi D, Zhang J, Wang M, Jing X, Humphry-Baker R, Wang P, Zakeeruddin SM, Grätzel M (2008) J Am Chem Soc 130:10720–10728
Sauvage F, Decoppet JD, Zhang M, Zakeeruddin SM, Comte P, Nazeeruddin MK, Wang P, Grätzel M (2011) J Am Chem Soc 133:9304–9309
Papageorgiou N, Maier WE, Grätzel M (1997) J Electrochem Soc 144:876–884
Murakami TN, Ito S, Wang Q, Nazeeruddin MK, Bessho T, Cesar I, Liska P, Humphry-Baker R, Comte P, Pechy P, Grätzel M (2006) J Electrochem Soc 153:A2255–A2261
Zukalova M, Zukal A, Kavan L, Nazeeruddin MK, Liska P, Grätzel M (2005) Nano Lett 5:1789–1792
Rothenberger G, Fitzmaurice D, Grätzel M (1992) J Phys Chem 96:5983–5986
Redmond G, Fitzmaurice D (1993) J Phys Chem 97:1426–1430
Kavan L, Stoto T, Grätzel M, Fitzmaurice D, Shklover V (1993) J Phys Chem 97:9493–9498
Zukalova M, Prochazka J, Zukal A, Yum JH, Kavan L (2008) Inorg Chim Acta 361:656–662
Fantacci S, De Angelis F (2011) Coord Chem Rev 2704–2726
Yu Q, Wang Y, Yi Z, Zu N, Zhang J, Zhang M, Wang P (2010) ACS Nano 4:6032–6038
Nazeeruddin MK, Kay A, Rodicio I, Humphry-Baker R, Mueller E, Liska P, Vlachopolous N, Grätzel M (1993) J Am Chem Soc 115:6382–6390
Morandeira A, Durante IL, O'Regan B, Martinez-Diaz MV, Fornelli A, Palomares E, Torres T, Durrant JR (2010) J Mater Chem 19:5016–5026
Yum JH, Jang SR, Humphry-Baker R, Grätzel M, Cid JJ, Torres T, Nazeeruddin MK (2008) Langmuir 24:5636–5640
De Angelis F, Fantacci S, Selloni A, Grätzel M, Nazeeruddin MK (2007) Nano Lett 7:3189–3195
Thomas AG, Flavell WR, Chatwin C, Rayner S, Tsoutsou D, Kumarasinghe AR, Brete D, Johal TK, Patel S, Purton J (2005) Surf Sci 592:159–168
Acknowledgments
This work was supported by the Academy of Sciences of the Czech Republic (contracts IAA 400400804 and KAN 200100801), Grant Agency of the Czech Republic (contract no. P108/12/0814), and the EC 7th FP project SANS (contract no. NMP-246124). B.L. thanks the National Research Foundation of Singapore (R279-000-276-272) for financial support. M.G. is very grateful to the European Research Council (ERC) for supporting his research under the ERC-2009-AdG grant no 247404 MESOLIGHT.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Laskova, B., Zukalova, M., Kavan, L. et al. Voltage enhancement in dye-sensitized solar cell using (001)-oriented anatase TiO2 nanosheets. J Solid State Electrochem 16, 2993–3001 (2012). https://doi.org/10.1007/s10008-012-1729-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-012-1729-0