
Abstract
In all organisms, carbamoylphosphate (CP) is a precursor common to the synthesis of arginine and pyrimidines. In Escherichia coli and most other Gram-negative bacteria, CP is produced by a single enzyme, carbamoylphosphate synthase (CPSase), encoded by the carAB operon. This particular situation poses a question of basic physiological interest: what are the metabolic controls coordinating the synthesis and distribution of this high-energy substance in view of the needs of both pathways? The study of the mechanisms has revealed unexpected moonlighting gene regulatory activities of enzymes and functional links between mechanisms as diverse as gene regulation and site-specific DNA recombination. At the level of enzyme production, various regulatory mechanisms were found to cooperate in a particularly intricate transcriptional control of a pair of tandem promoters. Transcription initiation is modulated by an interplay of several allosteric DNA-binding transcription factors using effector molecules from three different pathways (arginine, pyrimidines, purines), nucleoid-associated factors (NAPs), trigger enzymes (enzymes with a second unlinked gene regulatory function), DNA remodeling (bending and wrapping), UTP-dependent reiterative transcription initiation, and stringent control by the alarmone ppGpp. At the enzyme level, CPSase activity is tightly controlled by allosteric effectors originating from different pathways: an inhibitor (UMP) and two activators (ornithine and IMP) that antagonize the inhibitory effect of UMP. Furthermore, it is worth noticing that all reaction intermediates in the production of CP are extremely reactive and unstable, and protected by tunneling through a 96 Å long internal channel.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
CP at the crossroad of arginine and pyrimidine synthesis
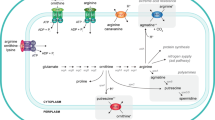
Carbamoylphosphate (CP) plays a dual metabolic role as it is required for both the de novo synthesis of arginine and pyrimidines (Fig. 1). The existence of a precursor common to both pathways was suggested by the isolation of one-step double auxotrophic E. coli mutants by RR Roepke, as quoted in (Tatum 1946). Ten years later, after extensive biochemical work, the molecule was identified as CP (Jones et al. 1955). In Escherichia coli, Salmonella typhimurium and related organisms, CP is synthesized from glutamine, two molecules of Mg2+ATP and bicarbonate by a single enzyme, carbamoylphosphate synthase (E.C. 6.3.5.5), encoded by the carAB operon (or its equivalent pyrA in Salmonella) (Piérard and Wiame 1964; Piérard et al. 1965; Abd-el-Al and Ingraham 1969; Abdelal and Ingraham 1975). This particular situation generates the need for a complex and tight control of CP production and its utilization for the de novo synthesis of arginine and pyrimidines in function of the cellular needs. In the arginine biosynthetic pathway, CP and ornithine are the substrates of ornithine carbamoyltransferase (OTCase) for the production of citrulline, which is further converted to arginine in two enzymatic steps (reviewed in Charlier and Glansdorff 2004). In the first committed step of pyrimidine biosynthesis, CP and aspartate are condensed into carbamoylaspartate, a reaction catalyzed by aspartate transcarbamylase (ATCase) (Lipscomb and Kantrowitz 2012), a paralog of OTCase (Labedan et al. 1999). A similar situation prevails in Salmonella typhimurium (Abdelal and Ingraham 1975), Serratia marcescens (Crane and Abdelal 1980), Pseudomonas aeruginosa (Abdelal et al. 1983), Proteus mirabilis (Prozesky and Coetzee 1966), Neisseria gonorrhoeae (Shinners and Catlin 1982) and enteric bacteria in general (Cunin et al. 1986). However, other microorganisms such as the Gram-positive bacteria Bacillus subtilis (Paulus and Switzer 1979), Geobacillus stearothermophilus (Yang et al. 1997) and Lactobacillus plantarum (Nicoloff et al. 2000), the yeast Saccharomyces cerevisiae and fungi such as Neurospora crassa (Lacroute et al. 1965; Bernhardt and Davis 1972; Davis 1986) possess two specialized and distinctly regulated CPSases, one for each pathway. In S. cerevisiae and N. crassa, the pyrimidine-specific CPSase encoded by the URA2 gene is part of a multifunctional protein exhibiting CPSase and ATCase activity (Lue and Kaplan 1969; Davis and Woodward 1962; Finck et al. 1965). In addition, the bifunctional protein carries an inactive dihydroorotase (DHO)-like domain in the linker connecting the CPSase and ATCase domains (Souciet et al. 1982, 1989). In the multifunctional CAD (CPSase, ATCase, DHOase) protein of higher eukaryotes, the dihydroorotase domain is functional. In some organisms, including bacteria such as Pseudomonas aeruginosa, Bacillus licheniformis, Streptococcus faecalis and Lactobacillus species, but also protists, moulds and halophilic archaea, CP can also be produced from citrulline by a catabolic OTCase of the arginine deiminase pathway, in which CP is then further used to generate ATP and ammonia (reviewed in Leroy and Charlier 2017). In hyperthermophilic archaea such as Pyrococcus species, a carbamate kinase rather than a CPSase appears to be responsible for CP synthesis (Purcarea et al. 1996; Durbecq et al. 1997; Rámon-Maiques et al. 2000; Uriarte et al. 1999; Marina et al. 1999; Alcántara et al. 2000). For additional information on CP producing and consuming enzymes in various organisms, including humans, the reader is referred to a recent review by Shi et al. (2018).

Biosynthesis of arginine and pyrimidines. The names of structural genes encoding the enzymes catalyzing the different steps of de novo arginine and pyrimidine synthesis are indicated as follows: argA (N-acetylglutamate synthase), argB (N-acetylglutamate kinase), argC (N-acetylglutamylphosphate reductase), argD (N-acetylornithine transaminase), argE (N-acetylornithinase), argF and argI (ornithine transcarbamylase, two isoenzymes), argG (argininosuccinate synthetase), argH (argininosuccinase), carAB (carbamoylphosphate synthase), pyrBI (aspartate transcarbamylase with pyrB encoded catalytic and pyrI encoded regulatory subunit), pyrC (dihydroorotase), pyrD (dihydroorotate dehydrogenase), pyrE (orotate phophoribosyltransferase), pyrF (orotidine-5′-phosphate decarboxylase), pyrH (UMP kinase), pyrG (CTP-synthase)
Transcriptional regulation of the E. coli carAB operon
Tandem promoters direct transcription of the carAB operon
The synthesis of E. coli CPSase is predominantly regulated at the transcriptional level as indicated by an almost perfect correlation between the levels of mRNA and enzyme activity over a large repression–derepression range (Piérard et al. 1980). The small glutaminase and large synthase subunit of the heterodimeric enzyme are encoded by the carA and carB gene, respectively, which from an operon (Mergeay et al. 1974; Gigot et al. 1980; Crabeel et al. 1980). It was first observed that the synthesis of CPSase is partially repressed upon growth in the presence of an excess of either arginine or a pyrimidine (usually uracil), and nearly completely in the presence of both, a phenomenon that was described as “cumulative repression” (Piérard and Wiame 1964; Piérard et al. 1965). Later on, it was demonstrated that the operon is transcribed from two adjacent promoters (Fig. 2), with startpoints 67 nt apart, and that excess pyrimidines switches off the upstream promoter P1, whereas excess arginine represses the downstream promoter P2 but does not affect initiation at P1 (Piette et al. 1984; Bouvier et al. 1984). In the genomes of E. coli and S. typhimurium, the operon is not directly linked to any other arginine or pyrimidine gene or transcriptional regulator affecting its expression.

Sequence and outline of the E. coli carAB control region. P1 and P2 represent the tandem pair of promoters directing carAB transcription, with indication of their respective − 10 and − 35 promoter elements, and transcription initiation site(s) (arrows). Distances are indicated with respect to the start of P1 transcription. Boxed sequences represent binding sites for the various transcription factors and trigger enzymes modulating transcription initiation at the P1 and P2 promoters. The two ARG boxes constitute the binding site for hexameric arginine-bound ArgR. The PUR box is the target of dimeric guanine or hypoxanthine-bound PurR. The RUT box is the target of unliganded dimeric RutR. The IHF box is the binding site of heterodimeric integration host factor (IHF), a nucleoid-associated protein. PepA1 and PepA2 represent sites of tight contact with the hexameric trigger enzyme PepA as identified in DNase I footprinting. The approximately 230 bp long region that gets wrapped around PepA is underlined. Dam represents the GATC sequence that is methylated by deoxyadenosine methyltransferase, a reaction that is inhibited upon binding of PepA and/or IHF. Notice that the initiation codon for carA mRNA translation is TTG (in bold)
Regulation of P2 promoter activity by arginine-bound ArgR
In vivo and in vitro P2 is repressed (about 50-fold as measured in a P2 only fusion construct) in the presence of excess arginine by hexameric ArgR (Piérard et al. 1972; Lissens et al. 1980; Charlier et al. 1988, 1992). ArgR is the repressor of arginine biosynthesis in E. coli and belongs to the winged helix-turn-helix (wHTH) family of DNA-binding transcription factors (Lim et al. 1987; Maas 1994; Van Duyne et al. 1996; Sunnerhagen et al. 1997). A single hexameric arginine-bound ArgR molecule cooperatively binds to two 18 bp long imperfect palindromes (ARG boxes) separated by three bp that partially overlap the − 35 promoter element (Fig. 2) and contacts major and minor groove determinants of the P2 operator, all aligned on one face of the DNA helix (Charlier et al. 1992; Wang et al. 1998). Mutations altering the sequence of these ARG boxes alleviate repression of the carAB operon by arginine and reduce the in vitro binding affinity for ArgR (Roovers et al. 1988; Charlier et al. 1992; Wang et al. 1998). Binding of RNA polymerase (RNAP) and ArgR to the P2 promoter region was shown to be mutually exclusive (Charlier et al. 1988). In contrast, ArgR does not impede the binding of RNAP to the P1 promoter and transcription initiated from this upstream promoter can proceed even in the presence of excess arginine (Piette et al. 1984; Charlier et al. 1988). Interestingly, ArgR-mediated repression of P2 was found to be stronger upon silencing initiation at P1 (either by physiological repression or mutational inactivation), suggesting that the ArgR•P2 operator complex is destabilized by RNAP binding at P1 or transcription initiated from P1 (Charlier et al. 1988). A very similar situation prevails in S. typhimurium, where carAB transcription is equally initiated from two tandem promoters and P2 repressed by ArgR binding to a pair of adjacent ARG boxes (Kilstrup et al. 1988; Lu et al. 1992). Interestingly, attenuation control that is operative in the control of many amino acid biosynthetic pathways is not involved in the regulation of P2 activity and neither in regulation of any other gene or operon of arginine biosynthesis in E. coli (Cunin et al. 1983).
Regulation of transcription initiation at P1
In contrast to the rather simple control mechanism operating at P2, unraveling the regulatory mechanisms modulating P1 activity turned out to be particularly challenging, and in spite of the identification of numerous players, the picture is still incomplete. It was clearly established that excess pyrimidines switches off P1 transcription (Charlier et al. 1988), but a general regulator of pyrimidine biosynthesis does not exist in E. coli, where all genes of pyrimidine biosynthesis are non-coordinately regulated by a variety of mechanisms (reviewed in Turnbough and Switzer 2008). In contrast, numerous DNA-binding transcription factors, nucleoid-associated factors (NAPs) and trigger enzymes (enzymes with a second unlinked gene regulatory function) were found to interact with an approximately 350 bp region upstream of the P1 promoter (Fig. 2). Roovers et al. (1988) isolated a first trans-acting regulatory element, CarP, in a search for P1 derepressed mutants. In carP mutants, P1 activity is no longer repressed upon uracil supplementation of the medium and about twofold higher than in the isogenic wild-type strain grown on minimal medium (Roovers et al. 1988; Charlier et al. 2000). Surprisingly, CarP turned out to be identical to PepA (Charlier et al. 1995b), an important Mn2+-dependent aminopeptidase in E. coli (Vogt 1970), and XerB, an auxiliary protein involved in conjunction with ArgR (alias XerA) in site-specific resolution of ColE1 multimers into the monomeric constituents (Guathakurta and Summers 1995; Stirling et al. 1989; Alén et al. 1997). Subsequent work indicated that the catalytic activity of PepA in not required for transcriptional regulation and that the protein binds to the P1 control region of E. coli and S. typhimurium in vitro in the absence of any effector molecule, and in vivo affects the methylation status of a GATC site in the P1 control region (Charlier et al. 1995a, b, 2000). PepA is a 330 kDa large homohexameric molecule (dimer of trimers) of which the structure has been solved at 2.5 Å resolution (Sträter et al. 1999). Each subunit consists of two domains: a smaller N-terminal domain (residues 1–166) is connected via a 26 amino acid long α-helix to the large C-terminal domain (residues 193–503) that bears the catalytic site. PepA does not possess a canonical DNA-binding motif, but on the basis of structural data and mutant studies, the N-terminal domain was shown to play an important role in DNA binding. DNA is proposed to dock into a groove running on the surface of the C-terminal domain of the protein and is bound by patches of basic residues that constitute the major N-terminal DNA-binding determinants at the ends of the groove (Sträter et al. 1999; Charlier et al. 2000; Reijns et al. 2005). DNase I footprinting assays and atomic force microscopy of PepA•P1 operator DNA complexes of E. coli and S. typhimurium indicated the deformation of an ~ 230 bp large region, extending from position − 280 to − 60 with respect to the start of P1 transcription, by wrapping around a single hexameric PepA molecule (Fig. 2) (Charlier et al. 1995b; Minh et al. 2009). This region comprises the binding sites for two transcriptional regulators, RutR and PurR, which equally interact with the P1 operator (see below). DNA topology assays indicated that wrapping of the P1 operator DNA around hexameric PepA generates a positive toroidal supercoil (Nguyen Le Minh et al. 2016). In view of the ligand-independent DNA binding of PepA, its multifunctional character, and the structural deformations, PepA was originally supposed to play a merely architectural role in P1 regulation. However, recently single-round in vitro transcription assays clearly demonstrated that PepA is a repressor in its own right, and hence a trigger enzyme, that specifically inhibits transcription initiation at P1, and the associated topological change is an integral part of the regulatory process (Nguyen Le Minh et al. 2016).
Besides PepA, three other proteins directly bind to the P1 control region and participate in modulation of P1 activity: RutR, the master regulator of genes involved in pyrimidine degradation (Shimada et al. 2007), PurR, the regulator of purine metabolism (Rolfes and Zalkin 1988; Cho et al. 2011), and the nucleoid-associated and DNA-bending protein IHF (integration host factor). RutR is a member of the TetR family of allosteric transcriptional regulators and was originally identified as the transcriptional regulator of the rutABCDEFG operon encoding a novel pathway for pyrimidine utilization in E. coli (Loh et al. 2006). Subsequent genomic SELEX and ChIP chip experiments identified 20 RutR binding sites on the E. coli genome, among which the carAB control region exhibits the highest affinity for the regulator (Shimada et al. 2008). RutR is an activator of transcription initiation at P1 and this stimulatory effect is strongly reduced in uracil-supplemented medium (Shimada et al. 2007). DNase I footprinting and high-resolution contact mapping of RutR•DNA complexes identified a 30 bp region of interaction, far upstream of the P1 promoter (from -200 to -170; Fig. 2) that bears a RUT box (-177 to -192) (Shimada et al. 2007; Nguyen Ple et al. 2010). In contrast to previous work (Shimada et al. 2007), structural work, modeling and mutant studies established that uracil but not thymine is the physiologically relevant ligand that abolishes DNA binding (Nguyen Le Minh et al. 2015). As the PepA and RutR binding sites overlap, competition in the binding of PepA and RutR has been proposed but not proven yet (Shimada et al. 2007).
In both E. coli and S. typhimurium P1 is about three to fourfold down-regulated by excess purines. This effect was first observed in S. typhimurium (Lu et al. 1995) and later in E. coli, where the molecular mechanism was studied (Devroede et al. 2004). PurR-mediated repression of P1 activity is much lower than purine-dependent repression of purine biosynthetic genes, but comparable to PurR-mediated repression of several other genes, including pyrC (DHOase) and pyrD (DHODase, dihydroorotate dehydrogenase) of pyrimidine biosynthesis (Choi and Zalkin 1990; Wilson and Turnbough 1990). In the presence of either guanine or hypoxanthine, the physiological effectors of PurR, the regulator binds to a 16 bp PUR box centered around position − 128.5 upstream of the start of P1 transcription (Fig. 2) and bends the operator by ~ 97° (Devroede et al. 2004), comparable to the PurR-induced deformation of the purF operator (Schumacher et al. 1997). This upstream position of the PurR-binding site in the P1 operator is unusual in the PurR regulon, where the binding site mostly partially overlaps the promoter or exceptionally works as a roadblock from a promoter downstream binding site (He and Zalkin 1992). In vitro transcription assays with purified PurR and analyses of the purine/PurR effect in various cis- and trans-acting mutants indicated that liganded PurR is by itself unable to repress P1 activity (Devroede et al. 2004, 2006). Instead, PurR appears to rely on the PepA-induced remodeling of the P1 control region to exert its regulatory effect. As a consequence, PurR- and PepA-mediated repression of P1 are structurally and functionally coupled, which is unprecedented in the action of PurR.
Integration host factor (IHF), a heterodimeric protein of similar subunits encoded by the himA and hip (alias himD) genes, and first discovered as a host-encoded protein involved in site-specific integration of bacteriophage lambda (Miller et al. 1979) is a multifunctional DNA-bending NAP that shows some sequence specificity for DNA binding (Goodrich et al. 1990; Goosen and van de Putte 1995). IHF was shown to bind to a highly A + T rich stretch far upstream (− 324 to − 287) of the start of P1 transcription in E. coli and S. typhimurium (Charlier et al. 1993). It is the most upstream located binding site in the P1 operator yet identified (Fig. 2). IHF stimulates P1 activity in cells grown in minimal medium but potentiates the pyrimidine-specific repression in uracil-supplemented medium. IHF thus appears to exert antagonistic effects and is required for both maximal expression and full repression of P1 (Charlier et al. 1993). As also observed for PepA, binding of this abundant NAP affects the methylation status of a GATC site 106 bp upstream of the start of P1 transcription (Charlier et al. 1994, 1995a). However, inhibition of methylation of this site appears to be a mere passive consequence of protein binding and not an active component of the regulatory process, since mutants of the GATC site that cannot be methylated are not impaired in P1 regulation (Charlier et al. 1995b).
Fis (factor for inversion stimulation), another NAP, was equally proposed to be involved in regulation of carA expression (Bradley et al. 2007). Fis is the most abundant NAP during early exponential phase (> 50,000 molecules/cell), when E. coli cells are dividing rapidly, but its concentration decreases tremendously during stationary phase (Ali Azam et al. 1999). Fis is involved in nucleoid structuring through binding to A-/AT tracts, but its local positive or negative effects on gene regulation are not well-understood (Cho et al. 2008). DNA microarray analyses in wild-type and mutant fis E. coli in different growth stages indicated an approximately twofold regulation of carA expression by Fis and predicted the presence of four A + T rich binding sites for Fis in the carAB control region, of which three are upstream of P1 and one overlapping the P2 promoter (Bradley et al. 2007). However, even though there is in vitro binding of purified Fis protein to P1 and P2 operator fragments observed in electrophoretic mobility shift assays (EMSA) in our laboratory, these complexes appeared to be non-specific. Furthermore, comparative reporter gene expression studies performed with isogenic wild-type and fis deletion strains in exponential growth phase did not reveal significant differences in P1 and P2 promoter activity (Islam Emdadul 2011).
Finally, Kholti et al. (1998) gathered evidence indicating that PyrH (UMP kinase) directly participates in pyrimidine-specific modulation of P1 activity. Interestingly, E. coli UMP kinase is a homohexameric protein, whereas most other AMP, GMP and UMP kinases are small monomeric proteins that share significant sequence homology (Briozzo et al. 2005). Furthermore, the enzyme does not display significant sequence homology with known UMP kinases of eukaryotic origin (Liljelund et al. 1989; Wiesmüller et al. 1990). In E. coli, pyrH is an essential gene and UMP kinase a highly regulated enzyme (inhibited by UTP, activated by GTP) that controls the de novo synthesis of all other pyrimidine nucleotides (Serina et al. 1995) (Fig. 1). In the search of P1 derepressed mutants on uracil-supplemented medium, pyrH mutants bearing a single amino acid substitution and retaining a quasi-normal UMP kinase activity were found to be impaired in P1 regulation (Kholti et al. 1998). Overexpression of the UMP–CMP kinase gene of Dictyostelium discoideum in such a mutant resulted in elevated UMP kinase levels, but did not restore normal control of P1, indicating that the slight reduction in UMP kinase activity and the potential concomitant reduction in the pyrimidine nucleotide pool is not responsible for the regulatory deficiency (Kholti et al. 1998). In vitro DNA-binding assays (EMSA and DNase I footprinting) with purified PyrH in the presence/absence of potential ligands did not reveal direct binding of the trigger enzyme to the P1 control region. Rather, on basis of preliminary yeast two hybrid assays (Nguyen 1998) and the opposite charge distribution on the surface of PyrH and PepA, two hexameric trigger enzymes involved in regulation of P1 activity, PyrH was proposed to be attracted to the P1 control region by protein–protein interaction (Briozzo et al. 2005; Marco-Marín et al. 2005), but this hypothesis has not been further validated yet.
Besides and independent of the complex TF-dependent regulation of transcription initiation that is operative at rather high UTP levels (0.9–1.4 mM), P1 activity is also regulated by a reiterative transcription control mechanism when the UTP level is low (0.9–50 µM) (Han and Turnbough 1998). These very low intracellular levels of UTP are not characteristic of exponentially growing prototrophs, but may occur subsequent to a sudden shift from high to low pyrimidine-supplemented medium, and in pyrimidine auxotrophs grown under limiting pyrimidine supply. Reiterative transcription, also known as transcriptional slippage or RNAP stuttering, is the repetitive addition of the same nucleotide to the 3’-end of the nascent transcript and results from the slippage between the transcript and the template DNA in a homopolymeric sequence. The phenomenon has been observed with RNAPs from all domains of life and with viral enzymes as well (Jacques and Kolakofsky 1991; Xiong and Reznikoff 1993; Cheng et al. 2001). In the context of pyrimidine metabolism, UTP-dependent reiterative transcription was first discovered for the pyrBI, codBA and upp genes and operons of E. coli (Liu and Turnbough 1989; Qi and Turnbough 1995; Tu and Turnbough 1997) and later for the carAB operon, where the effect is less pronounced (approximately threefold and two to threefold lower than for pyrBI) (Han and Turnbough 1998). P1 transcription starts with a G-residue followed by a series of three U residues (5′-GUUUGC-3′) (Fig. 2). Weak base pairing between the nascent GUUU transcript and its complementary DNA template allows for reversible one-base slippage and in the presence of high UTP concentrations, an extra U residue may be added to the 3′-end. Repeated rounds of slippage and extension may result in transcripts bearing long runs of U residues (5′-GUUUUn, with n = 1 to > 30), which are not further elongated into productive full-length mRNA molecules (Han and Turnbough 1998). At low UTP levels, the probability of inserting an extra U residue is much lower and even if slippage occurs, correct repositioning of the GUUU transcript will allow normal template-dependent transcription elongation. The importance of the polymeric stretch in the process is underscored by the observation that single T to G or T to C substitutions that interrupt the run of three T residues in the non-template strand of the initially transcribed region of the P1 promoter abolish the reiterative transcription control (Han and Turnbough 1998). Furthermore, mutant studies performed with the P1 promoter and comparative studies with the pyrBI and galETKM operons revealed the importance for stuttering of the initiating nucleotide (with A resulting in more reiterative transcription than G) and of the spacing between the − 10 promoter region and the transcription start site (with an 8 bp spacer generating more stuttering than the more canonical 7 bp linker) (Han and Turnbough 2014).
Finally, the P1 promoter was also shown to be subject to stringent control by the alarmone ppGpp (Bouvier et al. 1984). In a relA+ strain induction of isoleucine starvation by addition of valine to the culture medium resulted in an important increase in the ppGpp concentration and a concomitant approximately twofold reduction of P1 activity, which is very similar to the effect of stringent control on expression of the pyrBI operon (Turnbough 1983). However, the latter was measured in vitro in the absence of the small protein DksA, which is known to enhance the effect of ppGpp. The in vivo effect might thus be even stronger. It is worth noticing that in agreement with many other stringent promoters, P1 exhibits a six bp G + C rich sequence stretch (GCCGCC) immediately following the Pribnow box and preceding the G start nucleotide of the P1 mRNA (Fig. 2) that might function as a discriminator box and high-energy barrier for the isomerization from the closed to the open promoter complex (Dalebroux and Swanson 2012).
E. coli CPSase: reaction intermediates and enzyme structure
All CPSases use one molecule of bicarbonate, two molecules of Mg2+ATP, and one molecule of either glutamine or ammonia to synthesize CP (Fig. 3) (Jones and Lipman 1960; Anderson and Meister 1965; Shi et al. 2018). The preferred and physiologically relevant nitrogen donor for E. coli CPSase, and by extension for all bacterial CPSases, is glutamine (Piérard and Wiame 1964; Cunin et al. 1986; Meister 1989) (Fig. 3). Sequence conservation and structural characteristics indicate that the enzyme is part of the class I amidotransferase family of enzymes (Raushel et al. 1999). The reaction with ammonia is possible but requires very high and likely physiologically irrelevant concentrations (Rubino et al. 1987).

Reaction scheme of the synthesis of carbamoylphosphate by E. coli CPSase using glutamine as nitrogen donor
E. coli CPSase consists of two subunits, a small glutaminase subunit of ~ 41,000 (382 aa residues) encoded by carA and a large carB encoded subunit of 118,000 (1073 aa residues), that catalyzes the synthesis of CP from ammonia and also carries all the effector binding sites (Trotta et al. 1971; Matthews and Anderson 1972; Trotta et al. 1974a, b) (Fig. 4). carB is clearly the result of a gene duplication event, which suggests that CPSase evolved from a more primitive enzyme exhibiting kinase activity (Nyunoya and Lusty 1983; Lawson et al. 1996). The two halves of this large subunit exhibit 39% amino acid sequence identity, are independently folded, and functionally equivalent (except for the regulatory effects) whereby each part fixes one of the two Mg2+ATP molecules required for the synthesis of one molecule of CP (Guy and Evans 1996; Guy et al. 1997; Stapleton et al. 1996; Javid-Majd et al. 1996; Thoden et al. 1997). Combined structural, biochemical and genetic work provided detailed insight into the organization of the different domains of the enzyme (Fig. 4). The large subunit of E. coli CPSase is composed of four major domains (Thoden et al. 1997, 1999b). The carboxyphosphate domain (residues 1–400) and the homologous carbamate phosphorylation domain (residues 553–933; carbamoyl phosphate synthetic unit) exhibit nearly the same overall tertiary fold and are related by an axis of twofold rotational symmetry. Remarkably, the active sites of these two phosphorylation domains are separated by nearly 40 Å. These two domains are connected by an additional domain (residues 401–552) that appears to be required for oligomerization of the enzyme. Finally, the very C-terminus (residues 933–1073) constitutes the allosteric domain to which the allosteric effectors of CPSase activity bind (Rubio et al. 1991; Cervera et al. 1996; Czerwinsky et al. 1995; Delannay et al. 1999; Holden et al. 1999; Thoden et al. 1999c, d). The small glutaminase subunit makes contact with the carboxyphosphate domain, to which ammonia is delivered, and the domain involved in oligomerization. Cys-269 and His-353 of the glutaminase subunit are two residues essential for the hydrolysis of glutamine that passes through a thioester intermediate (Miran et al. 1991; Khedouri et al. 1966; Pinkus and Meister 1972; Anderson and Carlson 1975; Rubino et al. 1986; Mullins et al. 1991; Thoden et al. 1998, 1999a; Huang and Raushel 1999; Rishavy et al. 2000). From the structure, it appears that the three active sites in the enzyme are located far apart but connected by an intramolecular tunnel of 96 Å that leads from the glutamine binding site on the small subunit over the carboxyphosphate site (about 45 Å away) all the way to the carbamate phosphorylation site (another ~ 40 Å long tunnel) (Thoden et al. 1997; Huang and Raushel 2000a, b; Huang et al. 2001; Kim et al. 2002; Kim and Raushel 2004a, b; Fan et al. 2008, 2009; Lund et al. 2010). It is proposed that tunneling of the highly unstable reaction intermediates (Fig. 3) through the interior of the enzyme plays a crucial role in their protection (Thoden et al. 1997; Huang et al. 2001). CP itself is also a very thermolabile (half-life of < 2 s at 100 °C) and potentially harmful metabolite, as its decomposition produces the highly toxic cyanate (Allen and Jones 1964; Wang et al. 2008). However, binding of CP as a substrate to the active site of the tributary OTCase and ATCase enzymes strongly reduces its rate of thermal decomposition (Wang et al. 2008). In E. coli, there are at present no direct indications of further protection of CP through channeling between CPSase and the major CP consuming enzymes. This is, however, different in thermophilic organisms, where thermal degradation of CP would appear even more problematic. In the hyperthermophilic archaea Pyrococcus furiosus and P. abyssi, growing optimally at 100 °C, there is evidence for a weak physical association between the carbamate kinase-like CPSase and either OTCase or ATCase into a multienzyme cluster in which CP is directly transferred from the site of its synthesis to the active site of the consuming enzymes (Legrain et al. 1995; Purcarea et al. 1999; Massant et al. 2002; Massant and Glansdorff 2004, 2005). Channeling of CP to ATCase has also been demonstrated in the hyperthermophilic bacterium Aquifex aeolicus (Purcarea et al. 2003). Finally, partial channeling of CP in mesophilic hosts has also been shown to occur within the multienzymatic pyrimidine-specific CPSase–ATCase enzyme from the lower eukaryotes S. cerevisiae and N. crassa, and the CAD enzyme of mammals (Belkaïd et al. 1988; Penverne et al. 1994; Williams et al. 1970, 1971; Christopherson and Jones 1980; Irvine et al. 1997; Serre et al. 1999).

Ribbon presentation of the structure of heterodimeric (αβ) E. coli carbamoylphosphate synthase (pdb-1JDB) [133]. The small, carA encoded, glutamine amidotransferase subunit (magenta), and each domain of the large, carB encoded, catalytic subunit are indicated in different colors: carboxyphosphate synthetic component (green), oligomerization domain (yellow), carbamoylphosphate synthetic component (blue), allosteric domain (red). The figure was prepared with PyMOL
Allosteric regulation of CPSase activity
E. coli CPSase is a key enzyme involved in the synthesis of pyrimidine nucleotides and arginine, and hence occupies a strategic position in the production of building blocks for the synthesis of nucleic acids and proteins. This unique situation is not only reflected in the transcriptional control of the carAB operon as described above, but equally in the control of the enzyme’s activity. E. coli CPSase is an allosteric enzyme that is activated by ornithine and IMP, and inhibited by UMP (Piérard 1966; Anderson and Marvin 1968; Trotta et al. 1974a). The activating role of ornithine is crucial in view of the implication of the unique CPSase in arginine biosynthesis (Fig. 1). In presence of arginine, the synthesis of N-acetylglutamate synthetase, the first enzyme of arginine synthesis starting from glutamate, is repressed by arginine-bound ArgR and its activity feedback inhibited by arginine (Charlier and Glansdorff 2004). As a consequence, all pathway intermediates are present in limiting concentrations in this situation. When the arginine pool becomes too low to ensure protein synthesis, both repression and feedback inhibition of N-acetylglutamate synthetase are lifted and the concentration of ornithine increases. However, further conversion of the latter into citrulline requires CP. Activation of CPSase by accumulating ornithine (and concomitant derepression of the P2 promoter) thus ensures sufficient CP supply for the synthesis of arginine in this condition. The antagonistic effects of UMP and IMP may contribute to a correct balance between the production of purine and pyrimidine nucleotides in the cell. All three effector molecules primarily exert their effect on CPSase activity by modifying the apparent affinity of the enzyme for Mg2+ATP by approximately one order of magnitude, hence modulating the saturation of the enzyme by its substrate (Piérard 1966; Anderson and Meister 1966; Braxton et al. 1992, 1996).
The structure of E. coli CPSase was determined in the presence of different substrates and effectors, hence the binding sites for ornithine, IMP and UMP could be located and they all bind the C-terminal allosteric domain of CPSase (Thoden et al. 1997, 1999b, c, d, 2004). Previous studies performed with analogs of UMP and IMP already indicated that these antagonistic nucleotide effectors may bind the same or overlapping sites, but that this site is distinct from the ornithine binding site (Boettcher and Meister 1981, 1982; Mora et al. 1999; Fresquet et al. 2000; Braxton et al. 1999). The binding of UMP and ornithine essentially affect the binding of the second Mg2+ATP molecule (Braxton et al. 1992, 1999). Furthermore, the binding of ornithine and IMP decreases the affinity of the enzyme for the inhibitor UMP and vice versa, the binding of UMP lowers its affinity for the activators (Anderson 1977; Robin et al. 1989; Rubio et al. 1991). However, the activation by ornithine completely dominates the effects of the nucleotides (Braxton et al. 1999; Delannay et al. 1999). The inhibition in the binding of IMP by UMP is due to a competition in the binding of these two nucleotides for a common binding site (Anderson 1977; Boettcher and Meister 1981, 1982; Braxton et al. 1999; Bueso et al. 1999). From the solution of a co-crystal structure at 2.1 Å resolution IMP is now known to bind at the C-terminal portion of a five-stranded parallel β-sheet formed by the residues Ser937 to Lys1073 (Thoden et al. 1999c). Mutant studies and photolabeling assays indicated that Thr977 and Lys993 are crucial residues for UMP-dependent inhibition and UMP binding, respectively (Czerwinsky et al. 1995; Cervera et al. 1996; Pierrat and Raushel 2002). Structural data indicate that ornithine binds at the interface between the allosteric and the carbamoylphosphate domains. The carboxylate group of ornithine lies within hydrogen-bonding distance to both the backbone amide group and the side chain hydroxyl group of Thr1042 (Thoden et al. 1997, 1999b). Sequence determination and biochemical characterization of carB mutants isolated by Mergeay et al. (1974) pinpointed the crucial role of this residue in the allosteric regulation of CPSase activity (Delannay et al. 1999). Substitution of Thr1042 by Ileu greatly lowers the capacity of ornithine activation, but the enzyme is still sensitive to UMP and IMP, although to a lower extent (Delannay et al. 1999; Rochera et al. 2002; Pierrat et al. 2002). Similarly, substitution of Ser948 by Phe results in an enzyme that is insensitive to UMP and IMP, but still activated by ornithine, though again to a reduced extent (Delannay et al. 1999). As Thr1042 and Ser948 are located in spatially distinct regions of the enzyme, it is evident that the mutations have coupled effects on the activation and inhibition pathways of enzyme regulation. For a full description or overview of the different residues and molecular interactions involved in the binding of the different effectors, the reader is referred to the structural papers on wild-type and mutant CPSases (Thoden et al. 1997, 1998, 1999b, c, d).
CPSase is fundamentally a heterodimer (αβ) but this “monomeric” form readily converts to dimeric (αβ)2 and tetrameric (αβ)4 species depending on the presence of effector molecules (Powers et al. 1980; Anderson 1986). Ornithine, Mg2+ATP and potassium, all activators of the enzymatic reaction, promote the formation of the tetrameric species, whereas the enzyme exists essentially as a heterodimer (αβ) in the presence of the inhibitor UMP (Kim and Raushel 2001; Mora et al. 2002). Mutant studies indicated that dimer formation relies essentially on interactions of regulatory domains, whereas tetramers are formed by interaction of two dimers across their oligomerization domain, but noteworthy oligomerization per se has no effect on the regulation of CPSase activity (Mora et al. 2002).
Conclusion
The unique position of CP as a precursor common to the biosynthesis of arginine and pyrimidines, combined with the observation that E. coli harbors only a single enzyme that produces all the CP for both pathways, raises the need for a complex regulation of its synthesis at both the level of enzyme synthesis and enzyme activity. Such a combination is desirable and even indispensable since regulation of enzyme synthesis and allosteric regulation of enzyme activity operate at a different time scale. Regulation of enzyme synthesis by inhibition of transcription initiation affects the first step of gene expression and hence avoids waste of energy in the production of unnecessary mRNA and protein molecules, whereas fast inhibition/activation of enzyme activity by allosteric effector molecules allows fast adaptation of CP production in function of the needs of both pathways. This is particularly important in the case of CP, a labile molecule that should not accumulate in the cell. CP decomposition would not only represent a waste of energy (two moles of ATP are required to produce one mole of CP) but also generates the highly toxic cyanate. At the transcriptional level, the dual role of CP in the E. coli metabolism is reflected in the differential control mechanisms of the tandem pair of promoters directing transcription of the carAB operon. The downstream promoter P2 is repressed by arginine-bound ArgR, whereas the upstream promoter P1 is regulated by various mechanisms and effectors from both the pyrimidine and purine biosynthetic pathways. This situation ensures that enough CP is synthesized for the arginine pathway in conditions where P1 is repressed and vice versa that enough CP is produced for de novo pyrimidine synthesis when P2 is shut down upon high arginine supply. The antagonistic effects of ornithine (activator) and UMP (inhibitor) on CPSase activity and the consumption of CP as a substrate by the two carbamoylating enzymes (OTCase, ATCase) involved in the synthesis of arginine and UMP further ensure this delicate balance between CP production and CP consumption in function of the cellular requirements for both pathways. Much of the reaction mechanisms and their regulation, the enzyme structure and the protection of the highly labile intermediates in the production of CP could be solved by a combination of enzymatic, biochemical, genetic and structural studies with wild-type and mutant enzymes. These studies also revealed the presence of three distantly situated active sites connected through a 96Å internal tunnel. The study of transcriptional regulation of the carAB operon has revealed a complex interplay of several allosteric DNA-binding transcription factors using effector molecules from three different pathways (arginine, pyrimidines, purines), nucleoid-associated factors (NAPs), and trigger enzymes. Furthermore, DNA topology changes, UTP-dependent RNAP stuttering and stringent control by the alarmone ppGpp are involved as well. In this respect, regulation of CP production is an interesting example of the combined use of different gene regulatory mechanisms and effector molecules to coordinate the synthesis of a single metabolite involved in two distinct pathways. It also illustrates the diversity, flexibility and versatility of bacterial regulatory mechanisms. However, even though numerous players have been identified in the control of carAB transcription and CPSase activity, the mechanisms are not fully elucidated and much remains to be discovered on their interplay. ‘Avis aux amateurs’.
References
Abdelal AT, Ingraham JL (1975) Carbamylphosphate synthetase from Salmonella typhimurium. Regulations, subunit composition, and function of the subunits. J Biol Chem 250:4410–4417
Abd-el-Al A, Ingraham JL (1969) Control of carbamyl phosphate synthesis in Salmonella typhimurium. J Biol Chem 244:4033–4038
Abdelal AT, Bussey L, Vickers L (1983) Carbamoylphosphate synthetase from Pseudomonas aeruginosa. Subunit composition, kinetic analysis and regulation. Eur J Biochem 129:697–702
Alcántara C, Cervera J, Rubio V (2000) Carbamate kinase can replace in vivo carbamoyl phosphate synthetase. Implications for the evolution of carbamoyl phosphate biosynthesis. FEBS Lett 484:261–264
Alén C, Sherratt DJ, Colloms SD (1997) Direct interaction of aminopeptidase A with recombination site DNA in Xer site-specific recombination. EMBO J 16:5188–5197
Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A (1999) Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol 181:6361–6370
Allen CM Jr, Jones ME (1964) Decomposition of carbamoylphosphate in aqueous solutions. Biochemistry 3:1238–1247
Anderson PM (1977) Binding of allosteric effectors to carbamyl-phosphate synthetase from Escherichia coli. Biochemistry 16:587–593
Anderson PM (1986) Carbamoyl-phosphate synthetase: an example of effects on enzyme properties of shifting an equilibrium between active monomer and active oligomer. Biochemistry 25:5576–5582
Anderson PM, Carlson JD (1975) Reversible reaction of cyanate with a reactive sulphydryl group at the glutamine binding site of carbamyl phosphate synthetase. Biochemistry 14:3688–3694
Anderson PM, Marvin SV (1968) Effect of ornithine, IMP, and UMP on carbamyl phosphate synthetase from Escherichia coli. Biochem Biophys Res Commun 32:928–934
Anderson PM, Meister A (1965) Evidence for an activated form of carbon dioxide in the reaction catalyzed by Escherichia coli carbamoylphosphate synthetase. Biochemistry 4:2803–2809
Anderson PM, Meister A (1966) Control of Escherichia coli carbamyl phosphate synthetase by purines and pyrimidine nucleotides. Biochemistry 5:3164–3169
Belkaïd M, Penverne B, Hervé G (1988) In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisae. 3. Catalytic and regulatory properties of carbamoylphosphate synthetase: channeling of carbamoylphosphate to aspartate transcarbamylase. Arch Biochem Biophys 262:171–180
Bernhardt SA, Davis RH (1972) Carbamoyl phosphate compartmentation in Neurospora: histochemical localization of aspartate and ornithine transcarbamoylases. Proc Natl Acad Sci USA 69:1868–1872
Boettcher B, Meister A (1981) Conversion of UMP, an allosteric inhibitor of carbamyl phosphate synthetase, to an activator by modification of the UMP ribose moiety. J Biol Chem 256:5977–5980
Boettcher B, Meister A (1982) Regulation of Escherichia coli carbamyl phosphate synthetase. Evidence for overlap of the allosteric nucleotide binding sites. J Biol Chem 257:13971–13976
Bouvier J, Patte JC, Stragier P (1984) Multiple regulatory signals in the control region of the Escherichia coli carAB operon. Proc Natl Acad Sci USA 81:4139–4143
Bradley MD, Beach MB, de Koning AP, Pratt TS, Osuna R (2007) Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology 153(Pt 9):2922–2940
Braxton BL, Mullins LS, Raushel FM, Reinhart GD (1992) Quantifying the allosteric properties of Escherichia coli carbamyl phosphate synthetase: determination of thermodynamic linked-function parameters in an ordered kinetic mechanism. Biochemistry 31:2309–2316
Braxton BL, Mullins LS, Raushel FM, Reinhart GD (1996) Allosteric effects of carbamoyl phosphate synthetase from Escherichia coli are entropy-driven. Biochemistry 35:11918–11924
Braxton BL, Mullins LS, Raushel FM, Reinhart GD (1999) Allosteric dominance in carbamoyl phosphate synthetase. Biochemistry 38:1394–1401
Briozzo P, Evrin C, Meyer P, Assairi L, Joly N, Bârzu O, Gilles AM (2005) Structure of Escherichia coli UMP kinase differs from that of other nucleoside monophosphate kinases and sheds new light on enzyme regulation. J Biol Chem 280:25533–25540
Bueso J, Cervera J, Fresquet V, Marina A, Lusty CJ, Rubio V (1999) Photoaffinity labeling with the activator IMP and site-directed mutagenesis of histidine 995 of carbamoyl phosphate synthetase from Escherichia coli demonstrate that the binding site for IMP overlaps with that for the inhibitor UMP. Biochemistry 38:3910–3917
Cervera J, Bendala E, Britton HG, Bueso J, Nassif Z, Lusty CJ, Rubio V (1996) Photoaffinity labeling with UMP of lysine 992 of carbamyl phosphate synthetase from Escherichia coli allows identification of the binding sites for the pyrimidine inhibitor. Biochemistry 35:7247–7255
Charlier D, Glansdorff N (2004) Biosynthesis of arginine and polyamines. EcoSal Plus. https://doi.org/10.1128/ecosalplus.3.6.1.10
Charlier D, Weyens G, Roovers M, Piette J, Bocquet C, Piérard A, Glansdorff N (1988) Molecular interactions in the control region of the carAB operon encoding Escherichia coli carbamoylphosphate synthetase. J Mol Biol 204:867–877
Charlier D, Roovers M, Van Vliet F, Boyen A, Cunin R, Nakamura Y, Glansdorff N, Piérard A (1992) Arginine regulon of Escherichia coli K-12. A study of repressor-operator interactions and of in vitro binding affinities versus in vivo repression. J Mol Biol 226:367–386
Charlier D, Roovers M, Gigot D, Huysveld N, Piérard A, Glansdorff N (1993) Integration host factor (IHF) modulates the expression of the pyrimidine-specific promoter of the carAB operons of Escherichia coli K-12 and Salmonella typhimurium LT2. Mol Gen Genet 237:273–286
Charlier D, Huysveld N, Roovers M, Glansdorff N (1994) On the role of the Escherichia coli integration host factor (IHF) in repression at a distance of the pyrimidine specific promoter P1 of the carAB operon. Biochimie 76:1041–1051
Charlier D, Gigot D, Huysveld N, Roovers M, Piérard A, Glansdorff N (1995a) Pyrimidine regulation of the Escherichia coli and Salmonella typhimurium carAB operons: CarP and integration host factor (IHF) modulate the methylation status of a GATC site present in the control region. J Mol Biol 250:383–391
Charlier D, Hassanzadeh G, Kholti A, Gigot D, Piérard A, Glansdorff N (1995b) carP, involved in pyrimidine regulation of the Escherichia coli carbamoylphosphate synthetase operon encodes a sequence-specific DNA-binding protein identical to XerB and PepA, also required for resolution of ColE1 multimers. J Mol Biol 250:392–406
Charlier D, Kholti A, Huysveld N, Gigot D, Maes D, Thia-Toong TL, Glansdorff N (2000) Mutational analysis of Escherichia coli PepA, a multifunctional DNA-binding aminopeptidase. J Mol Biol 302:411–426
Cheng Y, Dylla SM, Turnbough CL Jr (2001) A long T•A tract in the upp initially transcribed region is required for regulation of upp expression by UTP-dependent reiterative transcription in Escherichia coli. J Bacteriol 183:221–228
Cho BK, Knight EM, Barrett CL, Palsson BØ (2008) Genome-wide analysis of Fis binding in Escherichia coli indicates a causative role for A-/AT tracts. Genome Res 18:900–910
Cho BK, Federowicz SA, Embree M, Park YS, Kim D, Palsson BØ (2011) The PurR regulon in Escherichia coli K-12 MG1655. Nucleic Acids Res 39:6456–6464
Choi KY, Zalkin H (1990) Regulation of Escherichia coli pyrC by the purine regulon repressor protein. J Bacteriol 172:3201–3207
Christopherson RI, Jones ME (1980) The overall synthesis of L-5,6-dihydroorotate by multienzymatic protein pyr1-3 from hamster cells. Kinetic studies, substrate channeling and effects of inhibitors. J Biol Chem 255:11381–11395
Crabeel M, Charlier D, Weyens G, Feller A, Piérard A, Glansdorff N (1980) Use of gene cloning to determine polarity of an operon: genes carAB of Escherichia coli. J Bacteriol 143:921–925
Crane CJ, Abdelal AT (1980) Regulation of carbamoylphosphate synthesis in Serratia marcescens. J Bacteriol 143:588–593
Cunin R, Eckhardt T, Piette J, Boyen A, Piérard A, Glansdorff N (1983) Molecular basis for modulated regulation of gene expression in the arginine regulon of Escherichia coli K-12. Nucleic Acids Res 11:5007–5019
Cunin R, Glansdorff N, Piérard A, Stalon V (1986) Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev 50:314–352
Czerwinski RM, Mareya SM, Raushel FM (1995) Regulatory changes in the control of carbamoyl phosphate synthetase induced by truncation and mutagenesis of the allosteric binding domain. Biochemistry 34:13920–13927
Dalebroux ZD, Swanson MS (2012) ppGpp: magic beyond RNA polymerase. Nat Rev Microbiol 10:203–212
Davis RH (1986) Compartmental and regulatory mechanisms in the arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol Rev 50:280–313
Davis RH, Woodward VW (1962) The relationship between gene suppression and aspartate transcarbamylase activity in pyr-3 mutants of Neurospora. Genetics 47:1075–1083
Delannay S, Charlier D, Tricot C, Villeret V, Piérard A, Stalon V (1999) Serine 948 and threonine 1042 are crucial residues for allosteric regulation of Escherichia coli carbamoylphosphate synthetase and illustrate coupling effects of activation and inhibition pathways. J Mol Biol 286:1217–1228
Devroede N, Thia-Toong TL, Gigot D, Maes D, Charlier D (2004) Purine and pyrimidine-specific repression of the Escherichia coli carAB operon are functionally and structurally coupled. J Mol Biol 336:25–42
Devroede N, Huysveld N, Charlier D (2006) Mutational analysis of intervening sequences connecting the binding sites for integration host factor, PepA, PurR, and RNA polymerase in the control region of the Escherichia coli carAB operon, encoding carbamoylphosphate synthase. J Bacteriol 188:3236–3245
Durbecq V, Legrain C, Roovers M, Piérard A, Glansdorff N (1997) The carbamate kinase-like carbamoyl phosphate synthetase of the hyperthermophilic archaeon Pyrococcus furiosus, a missing link in the evolution of carbamoyl phosphate biosynthesis. Proc Natl Acad Sci USA 94:12803–12808
Fan Y, Lund L, Yang L, Raushel FM, Gao YQ (2008) Mechanism for the transport of ammonia within carbamoyl phosphate synthetase determined by molecular dynamics simulations. Biochemistry 47:2935–2944
Fan Y, Lund L, Shao Q, Gao YQ, Raushel FM (2009) A combined theoretical and experimental study of the ammonia tunnel in carbamoyl phosphate synthetase. J Am Chem Soc 131:10211–10219
Finck D, Suyama Y, Davis RH (1965) Metabolic role of the pyrimidine-3 locus of Neurospora. Genetics 52:829–834
Fresquet V, Mora P, Rochera L, Ramón-Maiques S, Rubio V, Cervera J (2000) Site-directed mutagenesis of the regulatory domain of Escherichia coli carbamoyl phosphate synthetase identifies crucial residues for allosteric regulation and for transduction of the regulatory signals. J Mol Biol 299:979–991
Gigot D, Crabeel M, Feller A, Charlier D, Lissens W, Glansdorff N, Piérard A (1980) Patterns of polarity in the Escherichia coli carAB gene cluster. J Bacteriol 143:914–920
Goodrich JA, Schwartz ML, McClure WR (1990) Searching for and predicting the activity of sites for DNA binding proteins: compilation and analysis of the binding sites for Escherichia coli integration host factor (IHF). Nucleic Acids Res 18:4993–5000
Goosen N, van de Putte P (1995) The regulation of transcription initiation by integration host factor. Mol Microbiol 16:1–7
Guhathakurta A, Summers D (1995) Involvement of ArgR and PepA in the pairing of ColE1 dimer resolution sites. Microbiology 141(Pt 5):1163–1171
Guy HI, Evans DR (1996) Function of the major synthetase subdomains of carbamyl-phosphate synthetase. J Biol Chem 271:13762–13769
Guy HI, Bouvier A, Evans DR (1997) The smallest carbamoyl-phosphate synthetase. A single catalytic subdomain catalyzes all three partial reactions. J Biol Chem 272:29255–29262
Han X, Turnbough CL Jr (1998) Regulation of carAB expression in Escherichia coli occurs in part through UTP-sensitive reiterative transcription. J Bacteriol 180:705–713
Han X, Turnbough CL Jr (2014) Transcription start site sequence and spacing between the − 10 region and start site affect reiterative transcription-mediated regulation of gene expression in Escherichia coli. J Bacteriol 196:2912–2920
He B, Zalkin H (1992) Repression of Escherichia coli purB is by a transcriptional roadblock mechanism. J Bacteriol 174:7121–7127
Holden HM, Thoden JB, Raushel FM (1999) Carbamoyl phosphate synthetase: an amazing biochemical odyssey from substrate to product. Cell Mol Life Sci 56:507–522
Huang X, Raushel FM (1999) Deconstruction of the catalytic array within the amidotransferase subunit of carbamoyl phosphate synthetase. Biochemistry 38:15909–15914
Huang X, Raushel FM (2000a) An engineered blockage within the ammonia tunnel of carbamoyl phosphate synthetase prevents the use of glutamine as a substrate but not ammonia. Biochemistry 39:3240–3247
Huang X, Raushel FM (2000b) Restricted passage of reaction intermediates through the ammonia tunnel of carbamoyl phosphate synthetase. J Biol Chem 275:26233–26240
Huang X, Holden HM, Raushel FM (2001) Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem 70:149–180
Irvine HS, Shaw SM, Paton A, Carrey EA (1997) A reciprocal allosteric mechanism of efficient transfer of labile intermediates between active sites in CAD, the mammalian pyrimidine-biosynthetic multienzyme polypeptide. Eur J Biochem 247:1063–1073
Islam Emdadul Md (2011) On the role of Fis in the expression of the carAB operon of Escherichia coli. Dissertation, Vrije Universiteit Brussel
Jacques JP, Kolakofsky D (1991) Pseudo-templated transcription in prokaryotic and eukaryotic organisms. Genes Dev 5:707–713
Javid-Majd F, Stapleton MA, Harmon MF, Hanks BA, Mullins LS, Raushel FM (1996) Comparison of the functional differences for the homologous residues within the carboxy phosphate and carbamate domains of carbamoyl phosphate synthetase. Biochemistry 35:14362–14369
Jones ME, Lipmann F (1960) Chemical and enzymatic synthesis of carbamyl phosphate. Proc Natl Acad Sci USA 46:1194–1205
Jones ME, Spector L, Lipmann F (1955) Carbamyl phosphate, the carbamyl donor in enzymatic citrulline synthesis. J Am Chem Soc 77:819–820
Khedouri E, Anderson PM, Meister A (1966) Selective inactivation of the glutamine binding site of Escherichia coli carbamyl phosphate synthetase by 2-amino-4-oxo-5-chloropentanoic acid. Biochemistry 5:3552–3557
Kholti A, Charlier D, Gigot D, Huysveld N, Roovers M, Glansdorff N (1998) pyrH-encoded UMP-kinase directly participates in pyrimidine-specific modulation of promoter activity in Escherichia coli. J Mol Biol 280:571–582
Kilstrup M, Lu CD, Abdelal A, Neuhard J (1988) Nucleotide sequence of the carA gene and regulation of the carAB operon in Salmonella typhimurium. Eur J Biochem 176:421–429
Kim J, Raushel FM (2001) Allosteric control of the oligomerization of carbamoyl phosphate synthetase from Escherichia coli. Biochemistry 40:11030–11036
Kim J, Raushel FM (2004a) Access to the carbamate tunnel of carbamoyl phosphate synthetase. Arch Biochem Biophys 425:33–41
Kim J, Raushel FM (2004b) Perforation of the tunnel wall in carbamoyl phosphate synthetase derails the passage of ammonia between sequential active sites. Biochemistry 43:5334–5340
Kim J, Howell S, Huang X, Raushel FM (2002) Structural defects within the carbamate tunnel of carbamoyl phosphate synthetase. Biochemistry 41:12575–12581
Labedan B, Boyen A, Baetens M, Charlier D, Chen P, Cunin R, Durbecq V, Glansdorff N, Hervé G, Legrain C, Liang Z, Purcarea C, Roovers M, Sanchez R, Toong TL, Van de Casteele M, van Vliet F, Xu Y, Zhang YF (1999) The evolutionary history of carbamoyltransferases: a complex set of paralogous genes was already present in the last universal common ancestor. J Mol Evol 49:461–473
Lacroute F, Piérard A, Grenson M, Wiame JM (1965) The biosynthesis of carbamoyl phosphate in Saccharomyces cerevisiae. J Gen Microbiol 40:127–142
Lawson FS, Charlebois RL, Dillon J-AR (1996) Phylogenetic analysis of carbamoylphosphate synthetase genes: complex evolutionary history includes an internal duplication within a gene which can root the tree of life. Mol Biol Evol 13:970–977
Legrain C, Demarez M, Glansdorff N, Piérard A (1995) Ammonia-dependent synthesis and metabolic channeling of carbamoyl-phosphate in the hyperthermophilic archaeon Pyrococcus furiosus. Microbiology 141:1093–1099
Leroy F, Charlier D (2017) Arginine deiminase in microorganisms. In: D’Mello JPF (ed) The handbook of microbial metabolism of amino acids. CAB International Nosworthy Way, Wallingford. https://doi.org/10.1079/9781780647234.000
Liljelund P, Sanni A, Friesen JD, Lacroute F (1989) Primary structure of the S. cerevisiae gene encoding uridine monophosphokinase. Biochem Biophys Res Commun 165:464–473
Lim DB, Oppenheim JD, Eckhardt T, Maas WK (1987) Nucleotide sequence of the argR gene of Escherichia coli K-12 and isolation of its product, the arginine repressor. Proc Natl Acad Sci USA 84:6697–6701
Lipscomb WN, Kantrowitz ER (2012) Structure and mechanisms of Escherichia coli aspartate transcarbamoylase. Acc Chem Res 45:444–453
Lissens W, Cunin R, Kelker N, Glansdorff N, Piérard A (1980) In vitro synthesis of Escherichia coli carbamoylphosphate synthase: evidence for participation of the arginine repressor in cumulative repression. J Bacteriol 141:58–66
Liu CG, Turnbough CL Jr (1989) Multiple control mechanisms for pyrimidine-mediated regulation of pyrBI operon expression in Escherichia coli K-12. J Bacteriol 171:3337–3342
Loh KD, Gyaneshwar P, Markenscoff Papadimitriou E, Fong R, Kim KS, Parales R, Zhou Z, Inwood W, Kustu S (2006) A previously undescribed pathway for pyrimidine catabolism. Proc Natl Acad Sci USA 103:5114–5119
Lu CD, Houghton JE, Abdelal AT (1992) Characterization of the arginine repressor from Salmonella typhimurium and its interactions with the carAB operator. J Mol Biol 225:11–24
Lu CD, Katzif S, Abdelal AT (1995) Participation of the purine repressor in control of the carbamoylphosphate synthetase operon in Salmonella typhimurium. Mol Microbiol 17:981–988
Lue PF, Kaplan JG (1969) The aspartate transcarbamylase and carbamoyl phosphate synthetase of yeast: a multi-functional enzyme complex. Biochem Biophys Res Commun 34:426–433
Lund L, Fan Y, Shao Q, Gao YQ, Raushel FM (2010) Carbamate transport in carbamoyl phosphate synthetase: a theoretical and experimental investigation. J Am Chem Soc 132:3870–3878
Maas WK (1994) The arginine repressor of Escherichia coli. Microbiol Rev 58:631–640
Marco-Marín C, Escamilla-Honrubia JM, Rubio V (2005) First-time crystallization and preliminary X-ray crystallographic analysis of a bacterial-archaeal type UMP-kinase, a key enzyme in microbial pyrimidine biosynthesis. Biochim Biophys Acta 1747:271–275
Marina A, Alzari PM, Bravo J, Uriarte M, Barcelona B, Fita I, Rubio V (1999) Carbamate kinase: new structural machinery for making carbamoyl phosphate, the common precursor of pyrimidines and arginine. Protein Sci 8:934–940
Massant J, Glansdorff N (2004) Metabolic channeling of carbamoyl phosphate in the hyperthermophilic archaeon Pyrococcus furiosus: dynamic enzyme-enzyme interactions involved in the formation of the channeling complex. Biochem Soc Trans 32(Pt2):306–309
Massant J, Glansdorff N (2005) New experimental approaches for investigating interactions between Pyrococcus furiosus carbamate kinase and carbamoyltransferases, enzymes involved in the channeling of thermolabile carbamoyl phosphate. Archaea 1:365–373
Massant J, Verstreken P, Durbecq V, Kholti A, Legrain C, Beeckmans S, Cornelis P, Glansdorff N (2002) Metabolic channeling of carbamoyl phosphate, a thermolabile intermediate: evidence for physical interaction between carbamate kinase-like carbamoyl-phosphate synthetase and ornithine carbamoyltransferase from the hyperthermophilic Pyrococcus furiosus. J Biol Chem 277:18517–18522
Matthews SL, Anderson PM (1972) Evidence for the presence of two nonidentical subunits in the carbamyl phosphate synthetase of Escherichia coli. Biochemistry 11:1176–1183
Meister A (1989) Mechanism and regulation of the glutamine-dependent carbamyl phosphate synthetase: catalysis and regulation. Adv Enzyme Regul 16:289–315
Mergeay M, Gigot D, Beckmann J, Glansdorff N, Piérard A (1974) Physiology and genetics of carbamoylphosphate synthesis in Escherichia coli K-12. Mol Gen Genet 133:299–316
Miller HI, Kikuchi A, Nash HA, Weisberg RA, Friedman DI (1979) Site-specific recombination of bacteriophage lambda: the role of host gene products. Cold Spring Harb Symp Quant Biol 43(Pt 2):1121–1126
Minh PN, Devroede N, Massant J, Maes D, Charlier D (2009) Insights into the architecture and stoichiometry of Escherichia coli PepA•DNA complexes involved in transcriptional control and site-specific DNA recombination by atomic force microscopy. Nucleic Acids Res 37:1463–1476
Miran GS, Chang SH, Raushel FM (1991) Role of the four conserved histidine residues in the amidotransferase domain of carbamoyl phosphate synthetase. Biochemistry 30:7901–7907
Mora P, Rubio V, Fresquet V, Cervera J (1999) Localization of the site for the nucleotide effectors of Escherichia coli carbamoyl phosphate synthetase using site-directed mutagenesis. FEBS Lett 446:133–136
Mora P, Rubio V, Cervera J (2002) Mechanism of oligomerization of Escherichia coli carbamoyl phosphate synthetase and modulation by the allosteric effectors. A site-directed mutagenesis study. FEBS Lett 511:6–10
Mullins LS, Lusty CJ, Raushel FM (1991) Alterations in the energetics of the carbamoyl phosphate synthetase reaction by site-directed modification of the essential sulphydryl group. J Biol Chem 266:8236–8240
Nguyen M (1998) On the role of PyrH, PepA, and ArgR in the pyrimidine-specific regulation of the Escherichia coli carAB operon encoding carbamoylphosphate synthetase. Dissertation, Vrije Universiteit Brussel
Nguyen Le Minh P, de Cima S, Bervoets I, Maes D, Rubio V, Charlier D (2015) Ligand binding specificity of RutR, a member of the TetR family of transcription regulators in Escherichia coli. FEBS Open Bio 5:76–84
Nguyen Le Minh P, Nadal M, Charlier D (2016) The trigger enzyme PepA (aminopeptidase A) of Escherichia coli, a transcriptional repressor that generates positive supercoiling. FEBS Lett 590:1816–1825
Nguyen Ple M, Bervoets I, Maes D, Charlier D (2010) The protein-DNA contacts in RutR•carAB operator complexes. Nucleic Acids Res 38:6286–6300
Nicoloff H, Hubert JC, Bringel F (2000) In Lactobacillus plantarum, carbamoyl phosphate is synthesized by two carbamoyl-phosphate synthetases (CPS): carbon dioxide differentiates the arginine-repressed from the pyrimidine-regulated CPS. J Bacteriol 182:3416–3422
Nyunoya H, Lusty CJ (1983) The carB gene of Escherichia coli: a duplicated gene coding for the large subunit of carbamoyl-phosphate synthetase. Proc Natl Acad Sci USA 80:4629–4633
Paulus TJ, Switzer RL (1979) Characterization of pyrimidine-repressible and arginine-repressible carbamyl phosphate synthetases from Bacillus subtilis. J Bacteriol 137:82–91
Penverne B, Belkaïd M, Hervé G (1994) In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisiae. 4. The channeling of carbamoylphosphate to aspartate transcarbamylase and its partitioning in the pyrimidine and arginine pathways. Arch Biochem Biophys 309:85–93
Piérard A (1966) Control of the activity of Escherichia coli carbamoyl phosphate synthetase by antagonistic allosteric effectors. Science 154:1572–1573
Piérard A, Wiame JM (1964) Regulation and mutation affecting a glutamine dependent formation of carbamyl phosphate in Escherichia coli. Biochem Biophys Res Commun 15:76–81
Piérard A, Glansdorff N, Mergeay M, Wiame JM (1965) Control of the biosynthesis of carbamoyl phosphate in Escherichia coli. J Mol Biol 14:23–36
Piérard A, Glansdorff N, Yashphe J (1972) Mutations affecting uridine monophosphate pyrophosphorylase or the argR gene in Escherichia coli. Effects on carbamoyl phosphate and pyrimidine biosynthesis and on uracil uptake. Mol Gen Genet 118:235–245
Piérard A, Lissens W, Halleux P, Cunin R, Glansdorff N (1980) Role of transcriptional regulation and enzyme inactivation in the synthesis of Escherichia coli carbamoylphosphate synthase. J Bacteriol 141:382–385
Pierrat OA, Raushel FM (2002) A functional analysis of the allosteric nucleotide monophosphate binding site of carbamoyl phosphate synthetase. Arch Biochem Biophys 400:34–42
Pierrat OA, Javid-Majd F, Raushel FM (2002) Dissection of the conduit for allosteric control of carbamoyl phosphate synthetase by ornithine. Arch Biochem Biophys 400:26–33
Piette J, Nyunoya H, Lusty CJ, Cunin R, Weyens G, Crabeel M, Charlier D, Glansdorff N, Piérard A (1984) DNA sequence of the carA gene and the control region of carAB: tandem promoters, respectively controlled by arginine and the pyrimidines, regulate the synthesis of carbamoyl-phosphate synthetase in Escherichia coli K-12. Proc Natl Acad Sci USA 81:4134–4138
Pinkus LM, Meister A (1972) Identification of a reactive cysteine residue at the glutamine binding site of carbamyl phosphate synthetase. J Biol Chem 247:6119–6127
Powers SG, Meister A, Haschemeyer RH (1980) Linkage between self association and catalytic activity of Escherichia coli carbamyl phosphate synthetase. J Biol Chem 255:1554–1558
Prozesky OW, Coetzee JN (1966) Linked transductions in Proteus mirabilis. Nature 209:1262
Purcarea C, Simon V, Prieur D, Hervé G (1996) Purification and characterization of carbamoyl-phosphate synthetase from the deep-sea hyperthermophilic archaebacterium Pyrococcus abyssi. Eur J Biochem 236:189–199
Purcarea C, Evans DR, Hervé G (1999) Channeling of carbamoyl phosphate to the pyrimidine and arginine biosynthetic pathways in the deep sea hyperthermophilic archaeon Pyrococcus abyssi. J Biol Chem 274:6122–6129
Purcarea C, Ahuja A, Lu T, Kovari L, Guy HI, Evans DR (2003) Aquifex aeolicus aspartate transcarbamoylase, an enzyme specialized for the efficient utilization of unstable carbamoyl phosphate at elevated temperatures. J Biol Chem 278:52924–52934
Qi F, Turnbough CL Jr (1995) Regulation of codBA operon expression in Escherichia coli by UTP-dependent reiterative transcription and UTP-sensitive transcriptional start site switching. J Mol Biol 254:552–565
Ramón-Maiques S, Marina A, Uriarte M, Fita I, Rubio V (2000) The 1.5 Å resolution crystal structure of the carbamate kinase-like carbamoyl phosphate synthetase from the hyperthermophilic archaeon Pyrococcus furiosus, bound to ADP, confirms that this thermostable enzyme is a carbamate kinase, and provides insight into substrate binding and stability in carbamate kinases. J Mol Biol 299:463–476
Raushel FM, Thoden JB, Holden HM (1999) The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 38:7891–7899
Reijns M, Lu Y, Leach S, Colloms SD (2005) Mutagenesis of PepA suggests a new model for the Xer/cer synaptic complex. Mol Microbiol 57:927–941
Rishavy MA, Cleland WW, Lusty CJ (2000) 15N isotope effects in glutamine hydrolysis catalyzed by carbamyl phosphate synthetase: evidence for a tetrahedral intermediate in the mechanism. Biochemistry 39:7309–7315
Robin JP, Penverne B, Hervé G (1989) Carbamoyl phosphate biosynthesis and partition in pyrimidine and arginine pathways of Escherichia coli. In situ properties of carbamoyl-phosphate synthase, ornithine transcarbamylase and aspartate transcarbamylase in permeabilized cells. Eur J Biochem 183:519–528
Rochera L, Fresquet V, Rubio V, Cervera J (2002) Mechanism of allosteric modulation of Escherichia coli carbamoyl phosphate synthetase probed by site-directed mutagenesis of ornithine site residues. FEBS Lett 514:323–328
Rolfes RJ, Zalkin H (1988) Escherichia coli gene purR encoding a repressor protein for purine nucleotide synthesis. Cloning, nucleotide sequence, and interaction with the purF operator. J Biol Chem 263:19653–19661
Roovers M, Charlier D, Feller A, Gigot D, Holemans F, Lissens W, Piérard A, Glansdorff N (1988) carP, a novel gene regulating the transcription of the carbamoylphosphate synthetase operon of Escherichia coli. J Mol Biol 204:857–865
Rubino SD, Nyunoya H, Lysty C (1986) Catalytic domains of carbamyl phosphate synthetase. Glutamine hydrolyzing site of Escherichia coli carbamyl phosphate synthetase. J Biol Chem 261:11320–11327
Rubino SD, Nyunoya H, Lusty CJ (1987) In vivo synthesis of carbamyl phosphate from NH3 by the large subunit of Escherichia coli carbamyl phosphate synthetase. J Biol Chem 262:4382–4386
Rubio V, Cervera J, Lusty CJ, Bendala E, Britton HG (1991) Domain structure of the large subunit of Escherichia coli carbamoyl phosphate synthetase. Location of the binding site for the allosteric inhibitor UMP in the COOH-terminal domain. Biochemistry 30:1068–1075
Schumacher MA, Glasfeld A, Zalkin H, Brennan RG (1997) The X-ray structure of the PurR-guanine-purF operator complex reveals the contributions of complementary electrostatic surfaces and a water-mediated hydrogen bond to corepressor specificity and binding affinity. J Biol Chem 272:22648–22653
Serina L, Blondin C, Krin E, Sismeiro O, Danchin A, Sakamoto H, Gilles AM, Bârzu O (1995) Escherichia coli UMP-kinase, a member of the aspartokinase family, is a hexamer regulated by guanine nucleotides and UTP. Biochemistry 34:5066–5074
Serre V, Guy H, Penverne B, Lux M, Rotgeri A, Evans D, Hervé G (1999) Half of Saccharomyces cerevisiae carbamoyl phosphate synthetase produces and channels carbamoyl phosphate to the fused aspartate transcarbamoylase domain. J Biol Chem 274:23794–23801
Shi D, Caldovic L, Tuchman M (2018) Sources and fates of carbamyl phosphate: a labile energy-rich molecule with multiple facets. Biology. https://doi.org/10.3390/biology7020034
Shimada T, Hirao K, Kori A, Yamamoto K, Ishihama A (2007) RutR is the uracil/thymine-sensing master regulator of a set of genes for synthesis and degradation of pyrimidines. Mol Microbiol 66:744–757
Shimada T, Ishihama A, Busby SJ, Grainger DC (2008) The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions. Nucleic Acids Res 36:3950–3955
Shinners EN, Catlin BW (1982) Arginine and pyrimidine biosynthetic defects in Neisseria gonorrhoeae strains isolated from patients. J Bacteriol 151:295–302
Souciet JL, Hubert JC, Lacroute F (1982) Cloning and restriction mapping of the yeast URA2 gene coding for the carbamyl phosphate synthetase aspartate-transcarbamylase complex. Mol Gen Genet 186:385–390
Souciet JL, Nagy M, Le Gouar M, Lacroute F, Potier S (1989) Organization of the yeast URA2 gene: identification of a defective dihydroorotase-like domain in the multifunctional carbamoylphospate synthetase-aspartate transcarbamylase complex. Gene 79:59–70
Stapleton MA, Javid-Majd F, Harmon MF, Hanks BA, Grahmann JL, Mullins LS, Raushel FM (1996) Role of conserved residues within the carboxy phosphate domain of carbamoyl phosphate synthetase. Biochemistry 35:14352–14361
Stirling CJ, Colloms SD, Collins JF, Szatmari G, Sherratt DJ (1989) xerB, an Escherichia coli gene required for plasmid ColE1 site-specific recombination, is identical to pepA, encoding aminopeptidase A, a protein with substantial similarity to bovine lens leucine aminopeptidase. EMBO J 8:1623–1627
Sträter N, Sherratt DJ, Colloms SD (1999) X-ray structure of aminopeptidase A from Escherichia coli and a model from the nucleoprotein complex in Xer site-specific recombination. EMBO J 18:4513–4522
Sunnerhagen M, Nilges M, Otting G, Carey J (1997) Solution structure of the DNA-binding domain and model for the complex of multifunctional hexameric arginine repressor with DNA. Nat Struct Biol 4:819–826
Tatum EL (1946) Induced biochemical mutations in bacteria. Cold Spring Harb Symp Quant Biol 11:278–284
Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I (1997) Structure of carbamoyl phosphate synthetase: a journey of 96 Å from substrate to product. Biochemistry 36:6305–6316
Thoden JB, Miran SG, Phillips JC, Howard AJ, Raushel FM, Holden HM (1998) Carbamoyl phosphate synthetase: caught in the act of glutamine hydrolysis. Biochemistry 37:8825–8831
Thoden JB, Huang X, Raushel FM, Holden HM (1999a) The small subunit of carbamoyl phosphate synthetase: snapshots along the reaction pathway. Biochemistry 38:16158–16166
Thoden JB, Raushel FM, Benning MM, Rayment I, Holden HM (1999b) The structure of carbamoyl phosphate synthetase determined to 2.1 Å resolution. Acta Crystallogr D Biol Crystallogr 55(Pt 1):8–24
Thoden JB, Raushel FM, Wesenberg G, Holden HM (1999c) The binding of inosine monophosphate to Escherichia coli carbamoyl phosphate synthetase. J Biol Chem 274:22502–22507
Thoden JB, Wesenberg G, Raushel FM, Holden HM (1999d) Carbamoyl phosphate synthetase: closure of the B-domain as a result of nucleotide binding. Biochemistry 38:2347–2357
Thoden JB, Huang X, Kim J, Raushel FM, Holden HM (2004) Long-range allosteric transitions in carbamoyl phosphate synthetase. Protein Sci 13:2398–2405
Trotta PP, Burt ME, Haschemeyer RH, Meister A (1971) Reversible dissociation of carbamyl phosphate synthetase into a regulated synthesis subunit and a subunit required for glutamine utilization. Proc Natl Acad Sci USA 68:2599–2603
Trotta PP, Estis LF, Meister A, Haschemeyer RH (1974a) Self-association and allosteric properties of glutamine-dependent carbamyl phosphate synthetase. Reversible dissociation to monomeric species. J Biol Chem 249:482–489
Trotta PP, Pinkus LM, Haschemeyer RH, Meister A (1974b) Reversible dissociation of the monomer of glutamine-dependent carbamyl phosphate synthetase into catalytically active heavy and light subunits. J Biol Chem 249:492–499
Tu AH, Turnbough CL Jr (1997) Regulation of upp expression in Escherichia coli by UTP-sensitive selection of transcriptional start sites coupled with UTP-dependent reiterative transcription. J Bacteriol 179:6665–6673
Turnbough CL Jr (1983) Regulation of Escherichia coli aspartate transcarbamylase synthesis by guanosine tetraphosphate and pyrimidine ribonucleoside triphosphates. J Bacteriol 153:998–1007
Turnbough CL Jr, Switzer RL (2008) Regulation of pyrimidine biosynthetic gene expression in bacteria: repression without repressors. Microbiol Mol Biol Rev 72:266–300
Uriarte M, Marina A, Ramón-Maiques S, Fita I, Rubio V (1999) The carbamoyl-phosphate synthetase of Pyrococcus furiosus is enzymologically and structurally a carbamate kinase. J Biol Chem 274:16295–16303
Van Duyne GD, Ghosh G, Maas WK, Sigler PB (1996) Structure of the oligomerization and l-arginine binding domain of the arginine repressor of Escherichia coli. J Mol Biol 256:377–391
Vogt V (1970) Purification and properties of an aminopeptidase from Escherichia coli. J Biol Chem 245:4760–4769
Wang H, Glansdorff N, Charlier D (1998) The arginine repressor of Escherichia coli K-12 makes direct contacts to minor and major groove determinants of the operators. J Mol Biol 277:805–824
Wang Q, Xia J, Guallar V, Krilov G, Kantrowitz ER (2008) Mechanism of thermal decomposition of carbamoyl phosphate and its stabilization by aspartate and ornithine transcarbamoylases. Proc Natl Acad Sci USA 105:16918–16923
Wiesmüller L, Noegel AA, Bârzu O, Gerisch G, Schleicher M (1990) cDNA-derived sequence of UMP-CMP kinase from Dictyostelium discoideum and expression of the enzyme in Escherichia coli. J Biol Chem 265:6339–6345
Williams LG, Bernhardt S, Davis RH (1970) Copurification of pyrimidine-specific carbamyl phosphate synthetase and aspartate transcarbamylase of Neurospora crassa. Biochemistry 9:4329–4335
Williams LG, Bernhardt S, Davis RH (1971) Evidence for two discrete carbamyl phosphate pools in Neurospora. J Biol Chem 246:973–978
Wilson HR, Turnbough CL Jr (1990) Role of the purine repressor in the regulation of pyrimidine gene expression in Escherichia coli K-12. J Bacteriol 172:3208–3213
Xiong XF, Reznikoff WS (1993) Transcriptional slippage during the transcription initiation process at a mutant lac promoter in vivo. J Mol Biol 231:569–580
Yang H, Park SM, Nolan WG, Lu CD, Abdelal AT (1997) Cloning and characterization of the arginine-specific carbamoyl-phosphate synthetase from Bacillus stearothermophilus. Eur J Biochem 249:443–449
Acknowledgements
Work performed by the authors was supported by research grants from the Fonds Wetenschappelijk Onderzoek-Vlaanderen (Research Foundation-Flanders G.0429.06 and G.0321.13N) to DC and the Research Council of the Vrije Universiteit Brussel. PNLM was a post-doctoral fellow funded by the Research Foundation-Flanders.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: J. D. Wade.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Charlier, D., Nguyen Le Minh, P. & Roovers, M. Regulation of carbamoylphosphate synthesis in Escherichia coli: an amazing metabolite at the crossroad of arginine and pyrimidine biosynthesis. Amino Acids 50, 1647–1661 (2018). https://doi.org/10.1007/s00726-018-2654-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-018-2654-z