
Abstract
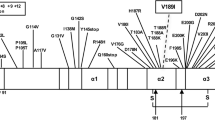
Genetic human prion diseases are autosomal dominant disorders associated with different mutations in the PRNP gene that are manifested as distinct clinical phenotypes. Here, we report a new pathogenic missense mutation (c.[643A>G], p.[I215V]) in the PRNP gene associated with three pathologically confirmed cases: two of Creutzfeldt–Jakob disease (CJD) and one of Alzheimer’s disease (AD) in two different families from the same geographical region in Spain. This mutation has not been found in any of more than 2,000 control cases studied. It represents a conservative amino acid change, and the same change is observed in the PRNP gene from other species. The two CJD cases were homozygous at codon 129 (M/M), but showed divergent clinical phenotypes with onset at ages 55 and 77 years and illness durations of 15 and 6 months, respectively. The postmortem neuropathological analysis of these cases showed homogeneous features compatible with CJD. Interestingly, the AD case (a brother of one of the CJD cases) was heterozygous at codon 129 (M/V). No familiar history was documented for any of the cases, suggesting a de novo mutation, or a partial, age-dependent penetration of the mutation, perhaps related to codon 129 status. This new mutation extends the list of known pathogenic mutations responsible for genetic CJD, reinforces the clinical heterogeneity of the disease, and advocates for the inclusion of PRNP gene examination in the diagnostic workup of patients with poorly classifiable dementia, even in the absence of family history.


Similar content being viewed by others

Abbreviations
- CJD:
-
Creutzfeldt–Jakob disease
- AD:
-
Alzheimer’s disease
- TSE:
-
Transmissible spongiform encephalopathies
- GSS:
-
Gerstmann–Sträussler–Scheinker syndrome
- FFI:
-
Fatal familial insomnia
References
Prusiner SB (1996) Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci 21:482–487
Weissmann C (1996) The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett 389:3–11
DeArmond SJ, Prusiner SB (2003) Perspectives on prion biology, prion disease pathogenesis, and pharmacologic approaches to treatment. Clin Lab Med 23:1–41
Wadsworth JD, Hill AF, Beck JA et al (2003) Molecular and clinical classification of human prion disease. Br Med Bull 66:241–254
Wadsworth JD, Collinge J (2011) Molecular pathology of human prion disease. Acta Neuropathol 121:69–77
Mead S (2006) Prion disease genetics. Eur J Hum Genet 14:273–281
Mastrianni JA (2010) The genetics of prion diseases. Genet Med 12:187–195
Lloyd S, Mead S, Collinge J (2011) Genetics of prion disease. Top Curr Chem 305:1–22
Pan KM, Baldwin M, Nguyen J et al (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90:10962–10966
Riek R, Hornemann S, Wider G et al (1996) NMR structure of the mouse prion protein domain PrP (121–231). Nature 382:180–182
Zahn R, Liu A, Lührs T et al (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97:145–150
Christen B, Hornemann S, Damberger FF et al (2009) Prion protein NMR structure from tammar wallaby (Macropus eugenii) shows that the beta2-alpha2 loop is modulated by long-range sequence effects. J Mol Biol 389:833–845
Christen B, Pérez DR, Hornemann S et al (2008) NMR structure of the bank vole prion protein at 20 degrees C contains a structured loop of residues 165–171. J Mol Biol 383:306–312
López Garcia F, Zahn R, Riek R et al (2000) NMR structure of the bovine prion protein. Proc Natl Acad Sci USA 97:8334–8339
Gossert AD, Bonjour S, Lysek DA et al (2005) Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA 102:646–650
Yoshida H, Terada S, Ishizu H et al (2010) An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology 30:159–164
Ghoshal N, Cali I, Perrin RJ et al (2009) Codistribution of amyloid beta plaques and spongiform degeneration in familial Creutzfeldt-Jakob disease with the E200K–129M haplotype. Arch Neurol 66:1240–1246
Jayadev S, Nochlin D, Poorkaj P et al (2011) Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 69:712–720
Heinemann U, Krasnianski A, Meissner B et al (2008) Novel PRNP mutation in a patient with a slow progressive dementia syndrome. Med Sci Monit 14:CS41–CS43
Kumar N, Boeve BF, Boot BP et al (2011) Clinical characterization of a kindred with a novel 12-octapeptide repeat insertion in the prion protein gene. Arch Neurol 68:1165–1170
Jellinger KA (2010) Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med 14:457–487
Calero O, Hortigüela R, Albo C et al (2009) Allelic discrimination of genetic human prion diseases by real-time PCR genotyping. Prion 3:146–150
Mead S, Mahal SP, Beck J et al (2001) Sporadic—but not variant—Creutzfeldt-Jakob disease is associated with polymorphisms upstream of PRNP exon 1. Am J Hum Genet 69:1225–1235
Calero O, Hortigüela R, Bullido MJ et al (2009) Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J Neurosci Methods 183:238–240
Cuadrado-Corrales N, Jiménez-Huete A, Albo C et al (2006) Impact of the clinical context on the 14-3-3 test for the diagnosis of sporadic CJD. BMC Neurol 6:25
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Goujon M, McWilliam H, Li W et al (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38:W695–W6999 (web service issue)
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Goldman JS, Miller BL, Safar J et al (2004) When sporadic disease is not sporadic: the potential for genetic etiology. Arch Neurol 61:213–216
Kovács GG, Puopolo M, Ladogana A et al (2005) EUROCJD. Genetic prion disease: the EUROCJD experience. Hum Genet 118:166–174
Goldfarb LG, Petersen RB, Tabaton M et al (1992) Fatal familial insomnia and familial Creutzfeldt–Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258:806–808
Brown P, Goldfarb LG, Kovanen J et al (1992) Phenotypic characteristics of familial Creutzfeldt–Jakob disease associated with the codon 178Asn PRNP mutation. Ann Neurol 31:282–285
Mitrová E, Belay G (2002) Creutzfeldt-Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol 46:31–39
Jarius C, Kovacs GG, Belay G et al (2003) Distinctive cerebellar immunoreactivity for the prion protein in familial (E200K) Creutzfeldt–Jakob disease. Acta Neuropathol 105:449–454
Gambetti P, Kong Q, Zou W et al (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239
Vollmert C, Windl O, Xiang W, Rosenberger A, Zerr I, Wichmann HE, Bickeböller H, Illig T, KORA group, Kretzschmar HA (2006) Significant association of a M129V independent polymorphism in the 5′ UTR of the PRNP gene with sporadic Creutzfeldt–Jakob disease in a large German case-control study. J Med Genet 43:e53
Kovács GG, Trabattoni G, Hainfellner JA et al (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249:1567–1582
Swietnicki W, Petersen RB, Gambetti P et al (1998) Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem 273:31048–31052
Rossetti G, Giachin G, Legname G et al (2010) Structural facets of disease-linked human prion protein mutants: a molecular dynamic study. Proteins 78:3270–3280
Shen L, Ji HF (2011) Mutation directional selection sheds light on prion pathogenesis. Biochem Biophys Res Commun 410:159–163
Meli M, Gasset M, Colombo G (2011) Dynamic diagnosis of familial prion diseases supports the β2-α2 loop as a universal interference target. PLoS One 6:e19093
Xu Z, Prigent S, Deslys JP et al (2011) Dual conformation of H2H3 domain of prion protein in mammalian cells. J Biol Chem 286:40060–40068
Agrimi U, Nonno R, Dell’Omo G et al (2008) Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog 4:e1000113
Eghiaian F, Daubenfeld T, Quenet Y et al (2007) Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc Natl Acad Sci USA 104:7414–7419
Gambetti P, Dong Z, Yuan J et al (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63:697–708
Zou WQ, Puoti G, Xiao X et al (2010) Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172
Beeri MS, Rapp M, Silverman JM et al (2006) Coronary artery disease is associated with Alzheimer disease neuropathology in APOE4 carriers. Neurology 66:1399–1404
Mahillo-Fernandez I, de Pedro-Cuesta J, Bleda MJ et al (2008) Surgery and risk of sporadic Creutzfeldt–Jakob disease in Denmark and Sweden: registry-based case-control studies. Neuroepidemiology 31:229–240
Acknowledgments
We thank the personnel from the Anatomic Pathology department of Hospital Clínico Universitario de Zaragoza (Zaragoza, Spain) for their assistance in the implementation of the autopsy of the three cases reported here, according to established protocols. We also thank all the physicians and epidemiological and clinical coordinators for their assistance and for notifying cases to the CJD Spanish Registry, as well as to F. Avellanal, J. Almazán, M. Ruiz, and E. Alcalde for their collaboration with the Spanish CJD surveillance system. This work was supported by grants FIS 05/0912 and PI11/03021 from the Acción Estratégica en Salud (Instituto de Salud Carlos III, Ministerio de Economía y Competitividad), the DGSP of the Spanish National Health Ministry (MC), and the Spanish CIBERNED network (JdP). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of interest
The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muñoz-Nieto, M., Ramonet, N., López-Gastón, J.I. et al. A novel mutation I215V in the PRNP gene associated with Creutzfeldt–Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol 260, 77–84 (2013). https://doi.org/10.1007/s00415-012-6588-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-012-6588-1