
Abstract
Congenital diaphragmatic hernia (CDH) is a major life-threatening cause of respiratory failure in the newborn. Recent data reveal the role of a retinoid-signaling pathway disruption in the pathogenesis of CDH. We describe the epidemiology and pathophysiology of human CDH, the metabolism of retinoids and the implications of retinoids in the development of the diaphragm and lung. Finally, we describe the existing evidence of a disruption of the retinoid-signaling pathway in CDH.
Similar content being viewed by others


Introduction
Congenital diaphragmatic hernia (CDH) remains the most life-threatening cause of severe respiratory failure in the term infant [1]. Its incidence is 1 in 2,500 births and accounts for about 8% of major congenital anomalies [2]. Approximately 80% are left-sided defects and the majority occurs as an isolated anomaly. CDH may, however, also be associated with a chromosomal abnormality including trisomy 13 and 18, Pallister–Killian syndrome and Fryns syndrome among others. Other associated malformations include congenital heart disease, spina bifida, hydrocephaly and urological anomalies [3]. Mortality in such cases may be as high as 90%, many fetuses succumbing in the antenatal period. Mortality in isolated CDH has been reported to range from 20 to 60% mainly because of the differences in case selection [4, 5]. Recent studies have emphasized the “hidden mortality” in CDH, showing survival of approximately 40% if all cases of CDH are included from the time of antenatal diagnosis [1, 5].
Despite remarkable progress in resuscitation and intensive care, the morbidity and mortality rates in CDH remain high due to severe pulmonary hypoplasia and persistent pulmonary hypertension resulting from the abnormal development of the pulmonary vascular bed. Postnatal therapies such as extracorporeal membrane oxygenation, inhaled nitric oxide, surfactant and high-frequency ventilation have had limited impact on prognosis and survivors of CDH commonly face considerable morbidity [6, 7]. Prenatal treatments have also been tried in CDH: open fetal diaphragmatic repair has not showed survival benefit [8] and a randomized controlled trial on fetoscopic tracheal occlusion has not shown to be beneficial [9]. Recently, a randomized study in the use of corticosteroids in late gestation in CDH has shown no benefit [10]. The disappointing effects of the studied treatments have stimulated studies aiming at revealing the exact etiopathology of CDH.
Pathophysiology
The classical hypothesis is that the clinical picture in CDH results from a failed closure of the pleuroperitoneal canals between the eighth and tenth week of gestation. It was believed that the associated pulmonary hypoplasia was caused by compression of the developing lung by herniation of abdominal viscera into the thorax [11].
Nitrofen (2,4-dichlorophenyl-p-nitropheniyl ether) is an herbicide that has been used for many years to create a teratogenic animal model of CDH [12]. Nitrofen has a low toxicity in adult rodents but its administration to pregnant rodents between day 8 and 11 after conception results in a high rate of CDH, associated pulmonary hypoplasia and pulmonary vascular abnormalities to their embryos, all remarkably similar to the human malformation [12]. The advantage of this model is that the defect is induced at the stage when the foregut has just separated into the esophagus and trachea, this model of CDH has given the opportunity of carefully studying the developmental anatomy of the lungs and diaphragm in CDH. Studies in the Nitrofen model of CDH have showed that hypoplasia of the lung develops early in gestation, before the normal closure of the diaphragm takes place [13]. These data led to the concept of a “dual-hit hypothesis”. This hypothesis explains pulmonary hypoplasia by two developmental insults. The first insult is caused by nitrofen and affects both lungs before diaphragm development. The second insult affects the ipsilateral lung after defective diaphragm development due to the compression of the lung by herniated viscera. Moreover, in a fibroblast growth factor 10 (FGF 10) null mutant mice which exhibit bilateral lung agenesis, Babiuk et al. [14] demonstrated that the diaphragm forms normally independently of lung development and defects in the diaphragm are induced in the absence of lung tissue. The same authors have demonstrated that the origin of the diaphragmatic defect lies in the amuscular mesenchymal precursor cells of the diaphragm, which are derived from the pleuroperitoneal folds. This theory is based on the observation that while migration of muscular precursors is not disturbed, a defect occurs in regions of the underlying mesenchymal substratum of the pleuroperitoneal folds [15].
Retinoids metabolism
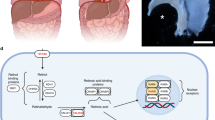
Retinoids are the family of molecules derived from vitamin A, which are essential for growth, development and differentiation [16]. Vitamin A is obtained from the diet in the form of retinyl esters present in animal meat or β-carotene present in vegetables. After absorption through the gut, retinyl esters are transported in chylomicrons to the liver for storage, where they are metabolized into retinol (Fig. 1). To meet tissue needs for retinol, livers secrete retinal bound to retinol-binding protein (RBP) into plasma, where it forms the main transporting complex with transthyretin. Circulating retinol binds to cell surface receptors and is internalized into target cells. Within the cytoplasm, retinol can be either oxidized to retinal or esterified to retinyl esters by lecitin retinol acetyltransferase (LRAT) for storage (Fig. 1). If converted to retinal, retinol is bound to cellular retinol binding protein I (CRBP-I) and is converted into retinal by retinol dehydrogenase (ROLDH) followed by a further dehydrogenation to retinoic acid (RA) using four cytosolic retinal dehydrogenases (RALDH). Although there are three known retinal dehydrogenases, RALDH1, RALDH2, and RALDH3, RALDH2 has a predominant role in generating retinoic acid. Retinoic acid is the biologically active metabolite of retinoids. Once retinoic acid has been synthesized in the cell, it enters the nucleus and binds to two ligand-activated nuclear transcription factors known as retinoic acid receptors (RARs) and retinoid X receptors (RXRs). There are three members of each class of receptor and these are known as α, β, and γ, thus giving a RARα, RARβ and RARγ and RXRα, RXRβ and RXRγ. To act on a responsive gene, one of the RARs heterodimerises with one of the RXRs. The RAR–RXR heterodimers are the functional units in transducing the retinoid signal at the gene level. Retinoic acid is further metabolized to inactive compounds by the action of Cyp26 enzyme, a newly recognized member of the cytochrome P450 family [16].

Overview of the retinoid-signaling pathway. After absorption through the gut, retinyl esters are transported in chylomicrons to the liver for storage, where they are metabolized into retinol. To meet the tissue needs for retinoids, the liver secretes retinol bound to the retinol binding protein (RBP) into the plasma, where it forms the main transporting complex with transthyretin (TTR). Within the cell cytoplasm, retinol is bound to cellular retinol biding protein (CRBP). Retinol can be either oxidized to retinal by an alcohol dehydrogenase (ROLDH) or esterified to retinyl esters by lecitin retinol acetyltransferase (LRAT) for storage. Retinal dehydrogenase (RALDH) then converts retinal to retinoic acid (RA). Retinoic acid exerts its biological effects through binding nuclear receptors, the retinoic acid receptors (RAR) heterodimerizes with the retinoid X receptors (RXR) to initiate transcription. In the cytoplasm, Cyp26 enzyme breaks down retinoic acid into inactive metabolites. It has been reported that LRAT activity could also be regulated by retinoic acid concentration
Retinoids and the development of lung and diaphragm
Mammalian lung development can be divided into five overlapping phases: embryonic, pseudoglandular, canalicular, saccular and alveolar period. Retinoids play an important role in each of the lung developmental stages. The human lung originates in the 3-week-old embryo as a ventral diverticulum that arises from the foregut. This endodermal bud grows caudally to form the trachea and then divides to form the two primary lung buds. By continuing dichotomous divisions, the two lung buds continue to form the bronchopulmonary segments of the mature lung. The structure of these buds consists of a columnar endoderm surrounded by splachnic mesoderm. By the sixth week of gestation, the pulmonary blood supply has connected to the developing heart [17]. Under conditions of acute maternal retinol deficiency, embryos display agenesis of the lung buds [18]. Retinoic acid biosynthesis and expression of RALDH2 are detected throughout the layers where the trachea and primitive lungs are forming [19]. CRBPs and retinoid receptors (RARβ, RXRα and β) are expressed in the early phases of lung formation [20, 21]. Using BMS493, a retinoic acid receptor antagonist (RARα, β and γ), mice embryos show no evidence of lung buds [22]. It has been demonstrated that a major role for retinoic acid in early lung morphogenesis is to selectively maintain mesodermal proliferation and induce fibroblast growth factor 10 expression in the foregut region in the initial budding of the lungs from the foregut [23].
The pseudoglandular stage of lung development occurs in the human between the seventh and sixteenth week of gestation [17]. During this period, conducting airways are formed by repeated dichotomous branching of the developing epithelium into the sorrounding mesenchyme. During this phase, the lung has a distinctly glandular appearance. By the 16th week of gestation, all bronchial airways have been formed. After this time, further growth occurs only by elongation and widening of existing airways and not by further branching [17]. As the developing lung begins this phase, RARs and RXRs continue to be expressed. RARα, RARγ, RXRα, RXRβ and RXRγ are present throughout the epithelium and mesenchyme at both proximal and distal sites. RARβ is present in proximal cells and in the distal mesenchyme but not in the distal epithelium [18]. RALDH2 expression is downregulated in regions of the lung that are undergoing budding and remains high in proximal regions of the lobes. Coincidently, a proximodistal gradient of CYP26, the enzyme that catabolizes retinoic acid, is established, with highest levels in distal buds. This may imply that during the pseudoglandular stage of lung development, retinoic acid may inhibit distal branching morphogenesis and act as a differentiating agent for proximal buds [18].
The canalicular phase of lung development takes place from the 16th to 26th week of gestation. This phase is characterized by the appearance of acini, consisting of an airway stem and a spray of short tubules arranged in a cluster surrounded by a covering of loose and thin mesenchyme. The mesenchyme becomes riddled with capillaries and is said to be “canalized”. The epithelial cells lining the tubules in the distal regions of the lung begin to flatten out and differentiate into type I and type II pneumocytes. At the end of this phase, gas exchange in the 26–28th week human can be supported. During this period, RARβ, RXRα and RXRβ are associated with epithelial cell differentiation and structural changes [18].
The saccular phase of lung development takes place from 24 weeks of gestation until term and is associated with striking changes in the appearance of the lung. There is a marked decrease of the prominence of interstitial tissue and a marked thinning of the airspace walls. Tissue projections into the distal airspace divide the distal airspaces into saccules. The cells lining the saccules of human fetal lung at this stage of development are recognizable type I and type II pneumocytes. Type II cells produce surfactant and are regarded as a lung progenitor cell population for type I cell [24]. During this period, retinoic acid stimulates proliferation of type II cells through an epithelial growth factor-mediated pathway [25]. Consistent results indicate that retinoic acid stimulates phospholipids synthesis, but there are considerable contradictory data on the effects of retinoic acid on surfactant protein production [18]. RALDH1 and RALDH2 are significantly upregulated during this period [26]. RALDH1 mRNA is expressed in the bronchial epithelium and alveolar interstitial tissue. RALDH2 mRNA is expressed in the bronchial epithelium and pleura, supporting the idea that RALDH2 could act as a local developmental signal to induce the distal formation of alveoli.
The alveolar period starts around the 36th week of gestation and continues postnatally, probably terminating by 18 or 24 months [27]. Alveolization starts with the appearance of crests along the sides of the saccular walls, which grow and divide the saccules into smaller units, the alveoli. The gas-exchange area therefore increases substantially while the mean alveolar size decreases. Two populations of fibroblasts are present in the alveolar wall. In one population there are abundant lipid droplets, the lipid interstitial fibroblasts, which synthesize and secrete retinoic acid [28]. The other population is the non-lipid interstitial fibroblasts. Together these cells are the major contributors to the synthesis of the extracellular matrix, which provides tensile strength and elasticity to the gas-exchanging surface [29]. Ultrastructural analysis has demonstrated that the lipid interstitial fibroblasts are in close contact with type II cells and that type II cells may be involved in the eruption of secondary septa [30]. Strong evidence for a role of retinoids in alveologenesis comes from a study from Massaro and Massaro [31]. These authors demonstrated a 50% increase in alveolar number along with a reduction in their size if retinoic acid was given to newborn mice. Moreover, recent data suggest that retinoic acid can induce alveolar regeneration in the adult mouse lung [32]. The function of the individual receptors during alveologenesis has been deduced from the phenotypes of the receptors knockouts. In postnatal RARγ −/− mice, the lungs contained less elastin and there was a decrease in alveolar wall volume density, alveolar surface area and number of alveoli [33]. RARα −/− postnatal mice, by contrast, had normal alveoli. But by day 50, alveolar volumes were higher, there were fewer alveoli and the alveolar surface area was lower [34]. This demonstrated that RARα plays a role in the continuing process of alveologenesis. The role of RARβ is even more surprising in that the alveolar volumes of RARβ −/− postnatal mice are smaller, there are more alveoli and the alveolar surface area is larger [35]. This suggests that RARβ is an endogenous inhibitor of alveolar septation.
According to the classical view, the diaphragm is formed from the septum transversum that separates the thoracic and abdominal cavities and leaves two pleuroperitoneal canals dorsally. By the 7th week of gestation, pleuroperitoneal folds extend from the lateral body wall and grow ventrally to fuse with the posterior margin of the septum transversum, thus sealing off the pleuroperitoneal canals. The definitive musculotendinous diaphragm incorporates muscle derivatives from four embryonic structures: the septum transversum, the pleuroperitoneal folds, the paraxial mesoderm of the body wall and the esophageal mesenchyme [36]. More recent embryological studies in the Nitrofen model of CDH have identified the pleuroperitoneal folds as the primary source of diaphragmatic musculature [37]. The pleuroperitoneal folds are derived from mesenchymal cells migrating from the somatic mesoderm. After that, muscle precursors migrate from the dermomyotome of cervical somites and localize in the pleuroperitoneal folds from where they migrate and from the lateral aspect of the septum transversum to the body wall and from the esophageal mesentery to the dorsal aspects of the crural region. The phrenic nerve makes contact with pleuroperitoneal folds, it trifurcates and extends intramuscular branches to the central, dorsolateral and crural areas of the developing diaphragm [38]. RALDH2 and RBP are strongly expressed in the pleuroperitoneal folds at postconceptional day 13 in rat [39, 40]. RARβ transcripts, but not RARγ transcripts are expressed in the rat diaphragm at postconception day 12.5 suggesting that RARβ could play a role in its formation [41].
Existing evidence of the role of retinoids in CDH
The first evidence linking retinoids with CDH came from examination of pups born to rat dams bred on a Vitamin A deficient diet. Diaphragmatic hernias were present in 25–70% of the offspring of Vitamin A deficient dams, the majority of them being right sided CDH [42, 43]. The rate of herniation was decreased when Vitamin A was introduced into the diet, being totally prevented if Vitamin A was given on days 10th and 11th of gestation [42].
Studies on retinoic acid receptors double knockout mice have demonstrated that some offspring have CDH. Interestingly, RAR αβ2 double mutant had right-sided defects, whereas RAR αβ2+/− double mutant had left-sided defects [41].
To date, there is only one clinical study linking retinoids to CDH [44]. Retinol and RBP were measured in 11 healthy newborns and in 11 CDH affected newborns. Both markers were decreased by 50% in cord blood of newborns with CDH when compared to newborns without CDH. Conversely, both retinol and RBP were significantly increased in the mothers of affected infants suggesting a possible deterioration of retinol transport across the placenta [44]. There is an ongoing multicenter international study aiming at measuring the retinoids state in mothers and newborn with CDH in a large cohort of patients (communication at the Congenital Diaphragmatic Hernia Work Study Group meeting, Houston, October 2007).
The largest evidence for the role of retinoids in CDH comes from the nitrofen model of CDH. The pathogenic mechanism of nitrofen, long unknown, has recently been clarified by Noble et al. [45]. These authors tested the three hypothesis regarding nitrofen’s mechanism of action: they examined the interaction of nitrofen with various aspects of the retinoid signaling pathway, examined the interaction of nitrofen and vitamins A, C and E to test the hypothesis that nitrofen was functioning as an antioxidant to interfere with retinoid signaling. Finally, they studied the interaction of nitrofen and thyroid signaling to test the hypothesis that because there are stereochemical similarities between the structure of nitrofen and thyroid hormone, nitrofen could alter thyroid hormone and thyroid hormone receptor functions. They concluded that nitrofen induces a marked decrease in retinoic acid levels, most probably due to inhibition of RALDH function rather than expression, since RALDH mRNA levels were normal. Retinoic acid synthesis perturbations did not appear to involve oxidative effects, neither perturbations of thyroid hormone signaling according to these authors [45]. These findings were consistent with previous biochemical data showing nitrofen suppression of RALDH2 activity [40].
The hypothesis that nitrofen disrupts the retinoid-signaling pathway was first tested using genetically engineered mice that had the lacZ reporter gene linked to a retinoic response element (RARE). There was a pronounced suppression of RARE-lacZ expression by nitrofen in in vitro whole embryo culture and in in vivo mouse model [46]. Further, the nitrofen-induced suppression was antagonized by supplemental administration of retinoic acid. Thebaud et al. [47] demonstrated that coadministration of large doses of Vitamin A along with nitrofen can reduce the incidence of CDH in the offpring from 84 to 20–40% depending on the day Vitamin A was given. They also demonstrated that Vitamin A could attenuate lung hypoplasia. These results were later confirmed by other investigators [48, 49]. Moreover, Babiuk et al. [50] demonstrated that the incidence of CDH is dramatically reduced from 54 to 8% if retinoic acid is given to pregnant rats along with nitrofen. The partial reduction in the incidence of nitrofen-induced CDH by the administration of large doses of Vitamin A is consistent with the hypothesis that the decreased enzyme activity can be partially countered by increasing the amount of substrate. Administration of retinoic acid bypasses the enzymatic alteration and explains the marked increase in efficacy of CDH reduction [50].
Nakazawa et al. have studied in depth the effects of nitrofen on the retinoid-signaling pathway. They demonstrated that there is a downregulation of Cyp26 and LRAT, meaning that retinoic acid is decreased in nitrofen-induced hypoplastic lungs [51]. However, they also demonstrated that lung retinol storage is decreased whereas serum retinol levels were significantly higher in the nitrofen model of CDH. Moreover, gene expression of most of the downstream components of the retinoid-signalling pathway, except for RALDH2, were increased in this model, suggesting a possible feedback reaction to the deficiency of lung retinol and that nitrofen may act by interfering with the cellular uptake of retinol during lung morphogenesis, resulting in pulmonary hypoplasia in this model [52].
Retinoids have also been used as a possible treatment in the nitrofen model of CDH. We showed that retinoic acid improves nitrofen-induced hypoplastic lung growth using fetal lung rat explants [53]. These results suggested that retinoid pathway might be involved not only in the pathogenesis of the diaphragmatic defect but also in the pathogenesis of associated pulmonary hypoplasia. We have recently demonstrated by means of stereological measurements and DNA content quantification, that retinoic acid promotes pulmonary alveologenesis in vivo at the end of gestation in the nitrofen model of CDH [54]. Moreover, Sugimoto et al. [55] have demonstrated that this is probably due to acceleration of type I alveolar cell proliferation of the hypoplastic lung in the nitrofen model of CDH.
Collectively, all these data strongly imply that CDH is caused by an interference with the retinoid-signalling pathway. If these experimental data are demonstrated in the human, retinoids may be used as an alternative therapeutic approach aimed at improving lung growth antenatally.
References
Stege G, Fenton A, Jaffray B (2003) Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics 112(3 Pt 1):532–535
Torfs CP, Curry CJ, Bateson TF, Honore LH (1992) A population-based study of congenital diaphragmatic hernia. Teratology 46(6):555–565
Sweed Y, Puri P (1993) Congenital diaphragmatic hernia: influence of associated malformations on survival. Arch Dis Child 69(1 Spec No):68–70
Hosgor M, Tibboel D (2004) Congenital diaphragmatic hernia; many questions, few answers. Paediatr Respir Rev 5(Suppl A):S277–S282
Ontario Congenital Anomalies Study Group (2004) Apparent truth about congenital diaphragmatic hernia: a population-based database is needed to establish benchmarking for clinical outcomes for CDH. J Pediatr Surg 39(5):661–665
Ivascu FA, Hirschl RB (2004) New approaches to managing congenital diaphragmatic hernia. Semin Perinatol 28(3):185–198
Doyle NM, Lally KP (2004) The CDH Study Group and advances in the clinical care of the patient with congenital diaphragmatic hernia. Semin Perinatol 28(3):174–184
Harrison MR, Adzick NS, Bullard KM, Farrell JA, Howell LJ, Rosen MA, et al (1997) Correction of congenital diaphragmatic hernia in utero VII: a prospective trial. J Pediatr Surg 32(11):1637–1642
Harrison MR, Keller RL, Hawgood SB, Kitterman JA, Sandberg PL, Farmer DL et al (2003) A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med 349(20):1916–1924
Lally KP, Bagolan P, Hosie S, Lally PA, Stewart M, Cotten CM, et al (2006) Corticosteroids for fetuses with congenital diaphragmatic hernia: can we show benefit? J Pediatr Surg 41(4):668–674; discussion 668–674
Puri P (1994) Congenital diaphragmatic hernia. Curr Probl Surg 31(10):787–846
Mortell A, Montedonico S, Puri P (2006) Animal models in pediatric surgery. Pediatr Surg Int 22(2):111–128
Keijzer R, Liu J, Deimling J, Tibboel D, Post M (2000) Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol 156(4):1299–1306
Babiuk RP, Greer JJ (2002) Diaphragm defects occur in a CDH hernia model independently of myogenesis and lung formation. Am J Physiol Lung Cell Mol Physiol 283(6):L1310–L1314
Babiuk RP, Zhang W, Clugston R, Allan DW, Greer JJ (2003) Embryological origins and development of the rat diaphragm. J Comp Neurol 455(4):477–487
Blomhoff R, Blomhoff HK (2006) Overview of retinoid metabolism and function. J Neurobiol 66(7):606–630
DiFiore JW, Wilson JM (1994) Lung development. Semin Pediatr Surg 3(4):221–232
Maden M (2004) Retinoids in lung development and regeneration. Curr Top Dev Biol 61:153–189
Malpel S, Mendelsohn C, Cardoso WV (2000) Regulation of retinoic acid signaling during lung morphogenesis. Development 127(14):3057–3067
Dolle P, Fraulob V, Kastner P, Chambon P (1994) Developmental expression of murine retinoid X receptor (RXR) genes. Mech Dev 45(2):91–104
Dolle P, Ruberte E, Leroy P, Morriss-Kay G, Chambon P (1990) Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential pattern of transcription during mouse organogenesis. Development 110(4):1133–1151
Mollard R, Ghyselinck NB, Wendling O, Chambon P, Mark M (2000) Stage-dependent responses of the developing lung to retinoic acid signaling. Int J Dev Biol 44(5):457–462
Desai TJ, Malpel S, Flentke GR, Smith SM, Cardoso WV (2004) Retinoic acid selectively regulates Fgf10 expression and maintains cell identity in the prospective lung field of the developing foregut. Dev Biol 273(2):402–415
Adamson IY, Bowden DH (1975) Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab Invest 32(6):736–745
Nabeyrat E, Besnard V, Corroyer S, Cazals V, Clement A (1998) Retinoic acid-induced proliferation of lung alveolar epithelial cells: relation with the IGF system. Am J Physiol 275(1 Pt 1):L71–L79
Hind M, Corcoran J, Maden M (2002) Alveolar proliferation, retinoid synthesizing enzymes, and endogenous retinoids in the postnatal mouse lung. Different roles for Aldh-1 and Raldh-2. Am J Respir Cell Mol Biol 26(1):67–73
Burri PH (1997) Structural aspects of prenatal and postnatal development and growth of the lung. In: McDonald JA (ed) Lung growth and development. Marcel Dekker, New York, pp 1–35
Dirami G, Massaro GD, Clerch LB, Ryan US, Reczek PR, Massaro D (2004) Lung retinol storing cells synthesize and secrete retinoic acid, an inducer of alveolus formation. Am J Physiol Lung Cell Mol Physiol 286(2):L249–L256
McGowan SE, Torday JS (1997) The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu Rev Physiol 59:43–62
Sannes PL (1991) Structural and functional relationships between type II pneumocytes and components of extracellular matrices. Exp Lung Res 17(4):639–659
Massaro GD, Massaro D (1996) Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. Am J Physiol 270(2 Pt 1):L305–L310
Hind M, Maden M (2004) Retinoic acid induces alveolar regeneration in the adult mouse lung. Eur Respir J 23(1):20–27
McGowan S, Jackson SK, Jenkins-Moore M, Dai HH, Chambon P, Snyder JM (2000) Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am J Respir Cell Mol Biol 23(2):162–167
Massaro GD, Massaro D, Chambon P (2003) Retinoic acid receptor-alpha regulates pulmonary alveolus formation in mice after, but not during, perinatal period. Am J Physiol Lung Cell Mol Physiol 284(2):L431–L433
Massaro GD, Massaro D, Chan WY, Clerch LB, Ghyselinck N, Chambon P et al (2000) Retinoic acid receptor-beta: an endogenous inhibitor of the perinatal formation of pulmonary alveoli. Physiol Genomics 4(1):51–57
Larsen W (1997) Human Embryology, 2nd edn. Churchill Livingstone, New York
Allan DW, Greer JJ (1997) Embryogenesis of the phrenic nerve and diaphragm in the fetal rat. J Comp Neurol 382(4):459–468
Greer JJ, Allan DW, Martin-Caraballo M, Lemke RP (1999) An overview of phrenic nerve and diaphragm muscle development in the perinatal rat. J Appl Physiol 86(3):779–786
Bavik C, Ward SJ, Ong DE (1997) Identification of a mechanism to localize generation of retinoic acid in rat embryos. Mech Dev 69(1–2):155–167
Mey J, Babiuk RP, Clugston R, Zhang W, Greer JJ (2003) Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am J Pathol 162(2):673–679
Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, et al (1994) Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120(10):2749–2771
Wilson JG, Roth CB, Warkany J (1953) An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat 92(2):189–217
Andersen DH (1941) Incidence of congenital diaphragmatic hernia in the young of rats bred on a diet deficient in Vitamin A. Am J Dis Child 62:888–889
Major D, Cadenas M, Fournier L, Leclerc S, Lefebvre M, Cloutier R (1998) Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr Surg Int 13(8):547–549
Noble BR, Babiuk RP, Clugston RD, Underhill TM, Sun H, Kawaguchi R et al (2007) Mechanisms of action of the congenital diaphragmatic hernia-inducing teratogen nitrofen. Am J Physiol Lung Cell Mol Physiol 293(4):L1079–L1087
Chen MH, MacGowan A, Ward S, Bavik C, Greer JJ (2003) The activation of the retinoic acid response element is inhibited in an animal model of congenital diaphragmatic hernia. Biol Neonate 83(3):157–161
Thebaud B, Tibboel D, Rambaud C, Mercier JC, Bourbon JR, Dinh-Xuan AT et al (1999) Vitamin A decreases the incidence and severity of nitrofen-induced congenital diaphragmatic hernia in rats. Am J Physiol 277(2 Pt 1):L423–L429
Baptista MJ, Melo-Rocha G, Pedrosa C, Gonzaga S, Teles A, Estevao-Costa J, et al (2005) Antenatal vitamin A administration attenuates lung hypoplasia by interfering with early instead of late determinants of lung underdevelopment in congenital diaphragmatic hernia. J Pediatr Surg 40(4):658–665
Oshiro T, Asato Y, Sakanashi M, Ohta T, Sugahara K (2005) Differential effects of vitamin A on fetal lung growth and diaphragmatic formation in nitrofen-induced rat model. Pulm Pharmacol Ther 18(3):155–164
Babiuk RP, Thebaud B, Greer JJ (2004) Reductions in the incidence of nitrofen-induced diaphragmatic hernia by vitamin A and retinoic acid. Am J Physiol Lung Cell Mol Physiol 286(5):L970–L973
Nakazawa N, Takayasu H, Montedonico S, Puri P (2007) Altered regulation of retinoic acid synthesis in nitrofen-induced hypoplastic lung. Pediatr Surg Int 23(5):391–396
Nakazawa N, Montedonico S, Takayasu H, Paradisi F, Puri P (2007) Disturbance of retinol transportation causes nitrofen-induced hypoplastic lung. J Pediatr Surg 42(2):345–349
Montedonico S, Nakazawa N, Puri P (2006) Retinoic acid rescues lung hypoplasia in nitrofen-induced hypoplastic foetal rat lung explants. Pediatr Surg Int 22(1):2–8
Montedonico S, Sugimoto K, Felle P, Bannigan J, Puri P (2008) Prenatal treatment with retinoic acid promotes pulmonary alveologenesis in the nitrofen model of congenital diaphragmatic hernia. J Pediatr Surg 43(3):500–507
Sugimoto K, Takayasu H, Nakazawa N, Montedonico S, Puri P (2008) Prenatal treatment with retinoic acid accelerates type 1 alveolar cell proliferation of the hypoplastic lung in the nitrofen model of congenital diaphragmatic hernia. J Pediatr Surg 43(2):367–372
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Montedonico, S., Nakazawa, N. & Puri, P. Congenital diaphragmatic hernia and retinoids: searching for an etiology. Pediatr Surg Int 24, 755–761 (2008). https://doi.org/10.1007/s00383-008-2140-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-008-2140-x