
Abstract
Aims/hypothesis
Our recent studies suggest that activation of the wingless-type MMTV integration site (WNT) pathway plays pathogenic roles in diabetic retinopathy and age-related macular degeneration. Here we investigated the causative role of oxidative stress in retinal WNT pathway activation in an experimental model of diabetes.
Methods
Cultured retinal pigment epithelial cells and retinal capillary endothelial cells were treated with a lipid peroxidation product, 4-hydroxynonenal (HNE), and an antioxidant, N-acetyl-cysteine (NAC). In vivo, rats with streptozotocin-induced diabetes were treated by NAC for 8 weeks. Activation of the canonical WNT pathway was measured by TOPFLASH assay and by western blot analysis of WNT pathway components and a WNT target gene, Ctgf. Oxidative stress in the retina was evaluated by immunostaining of HNE and 3-nitrotyrosine.
Results
Levels of phosphorylated and total LDL receptor-related protein (LRP)6, and cytosolic β-catenin, as well as transcriptional activity of T cell factor (TCF)/β-catenin were significantly increased by HNE. The production of connective tissue growth factor (CTGF) was also upregulated by HNE. NAC blocked the WNT pathway activation induced by HNE. Furthermore, LRP6 stability was increased by HNE and decreased by NAC. Retinal levels of HNE and 3-nitrotyrosine were significantly increased in diabetic rats, compared with those in non-diabetic rats. In the same diabetic rat retinas, levels of LRP6, cytosolic β-catenin and CTGF were significantly increased. NAC treatment reduced HNE and 3-nitrotyrosine levels and attenuated the upregulation of LRP6, β-catenin and CTGF in diabetic rat retina.
Conclusions/interpretation
Lipid peroxidation products activate the canonical WNT pathway through oxidative stress, which plays an important role in the development of retinal diseases.
Similar content being viewed by others


Introduction
Lipid peroxidation refers to the oxidative degradation of lipids. Reactive oxygen species including superoxide anion radical (O −2 ), hydrogen peroxide (H2O2), hydroxyl radical (OH⋅), nitric oxide (NO⋅) and peroxynitrite (ONOO−⋅) are most commonly involved in the initiation of lipid peroxidation [1]. The most intensively investigated lipid peroxidation product is 4-hydroxynonenal (HNE), a major oxidation product of membrane lipids containing polyunsaturated n-6 acyl groups. HNE was initially identified as a toxic endproduct of lipid peroxidation, but later emerged as a signalling mediator, which is involved in cell signal transduction and regulation of gene expression [2]. Increasing evidence has shown that tissue and blood levels of HNE are higher in a number of human diseases, including chronic inflammation, atherosclerosis, neurodegenerative diseases, diabetes and different types of cancer. It has been suggested that excessive HNE production could interfere with normal signalling pathways and have a potential pathogenic role in these diseases [3]. Besides the polyunsaturated fatty acids, cholesterol may also undergo free radical-mediated oxidation. One of the most important products of cholesterol peroxidation is 7-ketocholesterol, which is a main component of oxidised LDL and responsible for the cytotoxicity of oxidised LDL [4].
The retina is highly susceptible to oxidative stress and lipid peroxidation, as it has high levels of polyunsaturated fatty acids, the highest oxygen consumption per gram of tissue in the body and is also exposed to light [5]. Diabetic retinopathy and age-related macular degeneration (AMD) are leading causes of blindness in the USA [6, 7]. Although several lines of evidence have shown that lipid peroxidation products are important contributors to development of these diseases [8–10], the precise molecular mechanism that mediates the signalling transduction is still unclear.
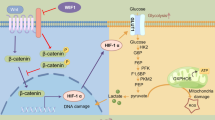
The WNT signalling pathway is versatile, regulating cell proliferation and differentiation, apoptosis, stem cell maintenance, angiogenesis, inflammation, fibrosis and carcinogenesis [11]. WNT signalling had been shown to be a key regulator of retinal development and has been implicated in certain retinal diseases [12]. WNT ligands are secreted cysteine-rich glycosylated proteins, which bind to a receptor complex comprising the frizzled receptors and the LDL receptor-related protein (LRP) 5 or 6. WNT ligand binding leads to phosphorylation and activation of LRP6, which is essential for WNT signalling. It also leads to inactivation of ‘destructive complex’, which is composed of glycogen synthase kinase-3β (GSK3β), axin and adenomatous polyposis coli. Inactivation of the ‘destructive complex’ prevents the proteosomal degradation of the transcriptional factor β-catenin and promotes its accumulation and nuclear translocation. Once β-catenin translocates into the nucleus, it associates with the T cell factor (TCF) and regulates transcription of the WNT target genes, including VEGF and CTGF [13–15].
Our previous studies have shown that the WNT pathway is activated in retina from human patients with diabetic retinopathy and in those from animal models of diabetes [16]. The WNT pathway is also activated in an animal model of AMD [17]. Moreover, blockade of WNT signalling attenuates retinal inflammation and neovascularisation in diabetic retinopathy and in animal models of AMD. These observations suggest that WNT pathway activation in diabetic retinopathy and AMD contributes to retinal inflammation and neovascularisation. The mechanism responsible for WNT pathway activation in these disease conditions is not clear.
The present study investigated the causative roles of oxidative stress and subsequent lipid peroxidation products in WNT pathway activation in AMD and diabetic retinopathy.
Methods
Materials and antibodies
HNE was purchased from Calbiochem (Madison, WI, USA). N-Acetyl-cysteine (NAC), hydrogen peroxide (H2O2), 7-ketocholesterol and cycloheximide were purchased from Sigma (St Louis, MO, USA). Normal human LDL and heavily oxidised glycated LDL (HOG-LDL) were isolated and prepared following a documented protocol [18]. Antibodies against β-catenin, total-LRP6 and connective tissue growth factor (CTGF) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies against phosphorylated-LRP6 (pLRP6) were from Cell Signaling (Danvers, MA, USA) and an anti-HNE adducts antibody was from Alpha Diagnostic (San Antonio, TX, USA). Antibodies for 3-nitrotyrosine and β-actin, and horseradish peroxidase-conjugated secondary antibodies were obtained from Abcam (Cambridge, MA, USA). The 2F1 monoclonal antibody was customer-generated through a contract with Protein-Tech (Chicago, IL, USA). Conditioned media containing wingless-type MMTV integration site family, member 3 (WNT3A) were prepared from mouse L cells (ATCC, Manassas, VA, USA) stably producing WNT3A. Control conditioned media were obtained from parental L cells.
Cell culture
ARPE19 cell line was purchased from ATCC and cultured according to the manufacturer’s protocol. Cells in the 15th to 30th passages were used for all experiments. Primary bovine retinal capillary endothelial cells (BRCEC) were isolated and cultured as described previously [19]. Cells in the fifth to sixth passages were used for all experiments.
Cell viability assay
ARPE19 cells in 96-well plates were treated with different stressors. Cell viability was measured using a kit (Cell Counting Kit-8; Dujindu, Rockville, MD, USA) according to the manufacturer’s protocol.
Dual-luciferase reporter assay
The TOPFLASH, FOPFLASH plasmids from the TCF Reporter Plasmid Kit (Millipore, Temecula, CA, USA) and renilla luciferase pRL-TK vectors were co-transfected into ARPE19 cells according to manufacturer’s protocol. TOPFLASH, FOPFLASH activity was measured using dual luciferase reporter system (Promega, Madison, WI, USA) and normalised to renilla luciferase activity.
Subcellular fractionation
Cells were fractionated using FractPrep (BioVision, Mountain View, CA, USA) following the manufacturer’s protocol.
Western blot analysis
Western blot analysis was performed as described previously [17].
Animals and induction of diabetes in rats
Female Brown Norway rats (8 weeks of age) were purchased from Harlan (Indianapolis, IN, USA). Care, use and treatment of all animals in this study were in strict agreement with the guidelines for the care and use of laboratory animals set forth by the University of Oklahoma. Experimental diabetes was induced as described previously [16].
Immunohistochemistry
Immunohistochemistry was performed as described previously [16]. Antibodies for 3-nitrotyrosine and HNE were used at a dilution of 1:200 and the antibody for β-catenin at a dilution of 1:300, with incubation overnight. After thorough washes with PBS, immunosignals were developed using a kit (Vectastain ABC; Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s protocol.
Statistical analysis
Data are presented as means ± SD. Comparisons were performed by two-tailed paired Student’s t test. A value of p < 0.05 was considered statistically significant.
Results
7-Ketocholesterol, H2O2 and HNE activated the canonical WNT pathway
H2O2 is one of the initiators of lipid peroxidation. 7-Ketocholesterol and HNE are the representative oxidation products of cholesterol and polyunsaturated acid, respectively. To define the sub-lethal concentration range of these three stimuli, ARPE19 cells were incubated separately with 7-ketocholesterol, H2O2 and HNE at various concentrations for 16 h. In the cells treated with 7-ketocholesterol, there was no cell viability change up to 10 μg/ml. At concentrations of 20–30 μg/ml, a slight decrease in cell viability was observed (Fig. 1a). In the H2O2-treated cells, cell viability was unaffected at up to 100 μmol/l. Cells incubated with 200 and 400 μmol/l of H2O2 showed 20% and 80% decreases in viability, respectively (Fig. 1b). However, in the HNE-treated cells no loss in cell viability was observed at 5 and 10 μmol/l (Fig. 1c). To exclude the possibility that the changes of the WNT pathway activity were caused by decreased number of viable cells, we chose concentrations that maintain most cells viable, allowing us to investigate the effects of these compounds on the canonical WNT pathway. The activities of the canonical WNT pathway were measured by TOPFLASH assay, an activity assay for luciferase reporter, driven by a promoter containing TCF/β-catenin-binding sites. As shown in Fig. 1d–f, the transcriptional activities of TCF/β-catenin were enhanced by 7-ketocholesterol, H2O2 and HNE in a dose-dependent manner. To confirm that the effects were dependent on TCF/β-catenin, we transfected the cells with the negative control FOPFLASH vector, which contains a scrambled DNA sequence of the TCF/β-catenin-binding sites. 7-Ketocholesterol, H2O2 and HNE treatments did not affect FOPFLASH activity. These results suggest that various lipid peroxidation products from different origins are causative factors for WNT signalling activation.

Effects of 7-ketocholesterol, H2O2 and HNE on cell viability and TOPFLASH activity. a ARPE19 cells were treated with various concentrations of 7-ketocholesterol (7-KC), (b) H2O2 and (c) HNE for 16 h. Cell viability was determined using a commercial kit and expressed as per cent of control cells. Values are mean ± SD, n = 3; *p < 0.05 and **p < 0.01. d ARPE19 cells were transfected for 8 h with the TOPFLASH or FOPFLASH plasmid and then treated with various concentrations of 7-ketocholesterol, (e) H2O2 and (f) HNE for another 16 h. TOPFLASH activity (white bars) and FOPFLASH activity (grey bars) were measured by luminometer and normalised to Renilla luciferase activity. Values are mean ± SD, n = 3; *p < 0.05 and **p < 0.01
The canonical WNT pathway was activated by HNE
To elucidate the mechanism by which lipid peroxidation products activate the canonical WNT pathway, HNE was chosen as a representative lipid peroxidation product for the mechanistic studies. As shown by western blot analysis, phosphorylation of LRP6, an initial step in WNT pathway activation, was increased by HNE in a concentration-dependent fashion (Fig. 2a, b). Total LRP6, a co-receptor of the WNT pathway, was also upregulated by HNE (Fig. 2a, c).

Activation of the canonical WNT pathway by HNE. ARPE19 cells were incubated with HNE for 6 h. a Levels of phosphorylated (p)LRP6 and total LRP6 were determined by western blot analysis using 50 μg of total proteins from each sample. b Quantification of pLRP6 and (c) total LRP6 by densitometry from three independent experiments, normalised to β-actin. d Levels of phosphorylated (p)GSK3β (Ser9) and (f) cytosolic β-catenin were determined by western blot analysis as above (a). e Quantification of pGSK3β (Ser9) and (g) β-catenin levels by densitometry from three independent experiments, normalised to β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. Values (b, c, e, g) are expressed as per cent of control cells, given as mean ± SD, n = 3; *p < 0.05 and **p < 0.01
In the canonical WNT pathway, β-catenin is phosphorylated by GSK3β in the absence of WNT ligands and then degraded in the cytosol. GSK3β activity is regulated by phosphorylation, e.g. phosphorylation at residue serine 9 (Ser9) is inhibitory to the kinase activity of GSK3β [20]. As shown in Fig. 2d, e, HNE induced an increase in phosphorylation of GSK3β at Ser9, indicating a decreased kinase activity of GSK3β. Consistently, cytosolic β-catenin levels were increased significantly in cells treated with HNE, compared with those in untreated cells (Fig. 2f, g), indicating stabilisation of β-catenin.
NAC attenuated HNE-induced activation of the canonical WNT pathway
It has been reported that HNE-induced Michael adduct formation can be blocked by pretreatment with thiol protectant [21]. NAC is an antioxidant as well as a thiol-protective agent. To determine whether the HNE-induced activation of WNT signalling is due to oxidative stress, we evaluated the effect of NAC on HNE-induced WNT pathway activation. As shown by TOPFLASH assay, the HNE-induced activity of the canonical WNT pathway was ameliorated by the NAC treatment, while FOPFLASH activity was not affected by these treatments (Fig. 3a). To elucidate the mechanism by which NAC blocked HNE-induced WNT activation, we measured phosphorylated and total LRP6, showing that exposure of cells to HNE caused an increase of phosphorylated and total LRP6 levels as compared with those in control cells. Pretreatment of the cells with NAC almost completely attenuated the induction of phosphorylated and total LRP6 by HNE (Fig. 3b–d). We also measured Lrp6 mRNA expression in cells exposed to HNE, and to HNE together with NAC. Real-time RT-PCR showed that there was no significant change of Lrp6 mRNA in the cells treated with HNE and HNE plus NAC (data not shown), suggesting that regulation of LRP6 levels by HNE did not occur at the transcription level.

Inhibition of HNE-induced WNT signalling by NAC. a ARPE19 cells were transfected with TOPFLASH or FOPFLASH plasmid for 8 h and then pretreated for 1 h with NAC (1 mmol/l) prior to challenge with HNE (10 μmol/l) for another 16 h. TOPFLASH activity (white bars) and FOPFLASH activity (grey bars) were measured by luminometer and normalised to Renilla luciferase activity. Values mean ± SD; n = 3; *p < 0.05 for HNE compared with control; † p < 0.05 for HNE+NAC compared with HNE. b ARPE19 cells were pretreated for 1 h with NAC (1 mmol/l) prior to challenge with HNE (10 μmol/l) for 6 h. Phosphorylated (p)LRP6 and total LRP6 levels were determined by western blot analysis using 50 μg of total proteins from each sample. The blot is representative of three independent experiments. c pLRP6 and (d) total LRP6 levels were quantified by densitometry and normalised to β-actin levels. Values are per cent of those in control cells, expressed as mean ± SD; n = 3; **p < 0.01 for HNE compared with control; † p < 0.05 and †† p < 0.01 for HNE+NAC compared with HNE. e ARPE19 cells were pretreated for 1 h with NAC (1 mmol/l) prior to challenge with HNE (10 μmol/l) for 16 h, followed by addition of cycloheximide (50 μg/ml) to block protein translation. Cells were collected at 0, 3, 5, 7 and 9 h following the addition of cycloheximide (Chx0). Total LRP6 protein levels at each time point were semi-quantified using western blot analysis, normalised to β-actin levels and are expressed (f) as per cent of the LRP6 levels prior to addition of cycloheximide. Blots (e) are representative of three independent experiments. Values (f) are mean ± SD; n = 3. Diamonds, control; squares, HNE; triangles, HNE + NAC
Next, we examined the protein stability of LRP6 in the cells treated with HNE and HNE plus NAC. We found that the half-life of LRP6 is approximately 3 h in untreated control cells. The HNE treatment significantly inhibited degradation of LRP6, prolonging its half-life. This stabilisation of LRP6 by HNE was reversed by NAC, suggesting that oxidative stress induces WNT pathway activation via stabilisation of LRP6 (Fig. 3e, f).
Effect of NAC on WNT ligand-induced WNT signalling
NAC is a glutathione precursor and increases glutathione levels [22]. A previous study reported that WNT or its related signalling proteins are the major targets of glutathione action [23]. Here we determined whether NAC affected the WNT pathway activation induced by WNT ligands. ARPE19 cells were exposed to WNT3A-conditioned medium with or without NAC for 6 h. Measurement of glutathione showed that NAC treatment induced a significant increase of glutathione levels (Fig. 4a). TOPFLASH assay showed that NAC blocked WNT3A-induced TOPFLASH activity (Fig. 4b). Furthermore, western blot analysis showed that WNT3A induced a 2.5-fold increase of LRP6 phosphorylation, which was attenuated by NAC (Fig. 4c, d).

Inhibitory effect of NAC on WNT3A-induced WNT signalling. a ARPE19 cells were pretreated for 1 h with NAC (1 mmol/l) prior to incubation with WNT3A-conditioned medium for 6 h. Glutathione (GSH) levels in the medium were measured using a commercial kit and expressed as per cent of those in control medium. Values are mean ± SD; n = 3; **p < 0.01. b ARPE19 cells were transfected for 8 h with the TOPFLASH or FOPFLASH plasmid, and then pretreated for 1 h with NAC (1 mmol/l) prior to incubation with WNT3A-conditioned medium for another 16 h. TOPFLASH activity (white bars) and FOPFLASH activity (grey bars) were measured by luminometer and normalised to Renilla luciferase activity. Values are mean ± SD; n = 3; **p < 0.01 for WNT3A compared with control; † p < 0.05 for WNT3A+NAC compared with WNT3A. c Phosphorylated (p)LRP6 and total LRP6 levels were determined with western blot analysis using 50 μg of total proteins from each sample. d pLRP6 was quantified by densitometry from three independent experiments, normalised to β-actin levels, and expressed as per cent of that in cells treated with control L medium. Values are mean ± SD; n = 3; **p < 0.01 for WNT3A compared with control; †† p < 0.01 for WNT3A+NAC compared with WNT3A
Level of CTGF was upregulated by HNE and inhibited by NAC
CTGF is one of the target genes regulated by the WNT pathway [15] and plays a pathogenic role in retinal diseases including diabetic retinopathy [24] and AMD [25]. To evaluate the pathogenic role of HNE in the regulation of WNT target genes in these diseases, the effects of HNE and NAC on CTGF levels were determined by western blot analysis, which showed that HNE induced a sixfold increase of CTGF levels, compared with control, while NAC downregulated CTGF production in the HNE-treated cells to a level similar to that in the untreated control (Fig. 5a, b). To distinguish whether HNE-induced CTGF production was through the canonical WNT pathway, we generated a monoclonal antibody specific to LRP6, 2F1, which binds to the E1E2 domains of LRP6 and blocks the canonical WNT pathway. ARPE19 cells were pretreated with the 2F1 antibody prior to HNE treatment. As shown by the TOPFLASH assay, HNE-induced WNT activation was inhibited significantly by the 2F1 antibody (Fig. 5c), indicating a blockade of HNE-induced activation of the canonical WNT pathway. Similarly, the CTGF production induced by HNE was also downregulated by the 2F1 antibody (Fig. 5d, e), indicating that the induction of CTGF by HNE was through the canonical WNT pathway.

CTGF was induced by HNE and attenuated by NAC. a ARPE19 cells were pretreated for 1 h with NAC (1 mmol/l) prior to challenge with HNE (10 μmol/l) for 6 h. CTGF levels were determined by western blot analysis using 50 μg of total proteins from each sample. CTGF was quantified (b) by densitometry from three independent experiments, normalised to β-actin levels and expressed as per cent of levels in control cells. Values mean ± SD; n = 3; **p < 0.01 for HNE compared with control); †† p < 0.01 for HNE+NAC compared with HNE. c ARPE19 cells were transfected for 8 h with the TOPFLASH or FOPFLASH plasmid and then pretreated with IgG or 2F1 (20 μg/ml) for 1 h prior to incubation with HNE (10 μmol/l) for another 16 h. TOPFLASH activity (white bars) and FOPFLASH activity (grey bars) were measured by luminometer and normalised to Renilla luciferase activity. Values are mean ± SD; n = 3; **p < 0.01 for HNE compared with control; †† p < 0.01 for HNE+2F1 compared with HNE. d ARPE19 cells were pretreated for 1 h with IgG or 2F1 (20 μg/ml) prior to incubation with HNE (10 μmol/l) for 6 h. CTGF levels were determined by western blot analysis using 50 μg of total proteins from each sample. CTGF was quantified (e) by densitometry from three independent experiments, normalised to β-actin levels and expressed as per cent of levels in control cells. Values are mean ± SD; **p < 0.01 for HNE compared with control; †† p < 0.01 for HNE+2F1 compared with HNE
β-Catenin was induced by HNE and HOG-LDL, and blocked by NAC treatment in endothelial cells
Dysfunction of endothelial cells is a key pathological change in the development of retinal diseases, especially in diabetic retinopathy [7]. We have previously shown that oxidised and glycated LDL is implicated in the initiation and development of diabetic retinopathy [26]. Previous studies in our group have shown that the canonical WNT pathway is activated in diabetic retinopathy in human patients and animal models of disease [16]. To determine the effect of lipid peroxidation products on the WNT pathway in endothelial cells, we measured the β-catenin level in BRCEC exposed to HNE and HOG-LDL (20 μg/ml). Western blot analysis showed that β-catenin levels were elevated in the cells exposed to HNE or HOG-LDL, compared with their respective control cells. NAC treatment significantly blocked the increases of β-catenin levels induced by HNE and HOG-LDL (Fig. 6a, b). These results demonstrate that the pathogenic role of lipid peroxidation products in diabetic retinopathy occurred partly through activation of the WNT pathway.

β-Catenin levels were increased by HNE and HOG-LDL, and decreased by NAC in BRCEC. a BRCEC were pretreated for 1 h with NAC (1 mmol/l) prior to challenge with HNE (10 μmol/l). Cytosolic β-catenin was measured by western blot analysis using 50 μg of proteins from each sample, normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and quantified in bar graph as per cent of levels in control. Values are mean ± SD; n = 3; **p < 0.01 for HNE compared with control; †† p < 0.01 for HNE+NAC compared with HNE. b BRCEC were treated with normal LDL (N-LDL) and HOG-LDL (20 μg/ml) for 12 h. β-Catenin levels were determined by western blot analysis using 50 μg of proteins from each sample, normalised to β-actin levels and expressed as per cent of those in control. Values are mean ± SD; n = 3; **p < 0.01 for HOG-LDL compared with N-LDL; † p < 0.05 for HOG+NAC compared with HOG-LDL
Causative effects of oxidative stress on the WNT pathway in the retina of diabetic rats
It is well documented that oxidative stress and lipid peroxidation products play important roles in diabetic retinopathy, while inhibitors of lipid peroxidation have beneficial effects in diabetic complications [9, 16]. A recent study reported that NAC treatment reduced free radicals and ameliorated early events of diabetic retinopathy in a streptozotocin-induced animal model of diabetes [27]. Our previous study reported that the WNT pathway is activated in the retina of streptozotocin-induced diabetic rats and Akita mice [16]. To establish the causative role of oxidative stress in WNT pathway activation in diabetes, we assessed oxidative stress and WNT pathway activities in the retina. Immunostaining of the retinal sections showed that immunosignals of HNE and 3-nitrotyrosine, another oxidative stress maker [28], were more intense in diabetic rats than in non-diabetic control rats, suggesting an increase in oxidative stress and subsequent lipid peroxidation in the retina of these rats. Correlating with the increased oxidative stress, the intensity of the β-catenin immunosignal was also increased in the same diabetic retinas. NAC treatment (2 months) of the diabetic rats significantly reduced HNE and 3-nitrotyrosine in the retina of the diabetic rats (Fig. 7a). Consistent with this, NAC treatment also decreased β-catenin levels in the diabetic retina, compared with those in untreated diabetic rat retinas. Western blot analysis confirmed that retinal levels of 3-nitrotyrosine, total LRP6, β-catenin and CTGF were upregulated in diabetic rats and reduced by NAC treatment (Fig. 7b), suggesting that oxidative stress is a causative factor in the canonical WNT pathway activation in diabetic retinopathy.

Effects of oxidative stress, lipid peroxidation products and NAC on the WNT pathway in the retina of diabetic rats. a Normal rats at 8 weeks of age received an intraperitoneal injection of streptozotocin (50 mg/kg in 10 mmol/l of citrate buffer; pH 4.5) after an overnight fast. Age-matched control rats received an injection of the citrate buffer alone for non-diabetic control (Non-DM). NAC (1.4 g/kg) was administered to diabetic groups (DM) in the drinking water for 8 weeks. The ocular sections were immunostained separately with antibodies for 3-nitrotyrosine, HNE and β-catenin using DAB staining. GCL, Ganglion cell layer; OPL, outer plexiform layer. b The eyecups were dissected, and levels of total LRP6, β-catenin and CTGF were determined by western blot analysis using 50 μg of total proteins from each sample. Each lane represents an individual animal. Wt, wild-type
Discussion
Our previous studies have shown that activation of the WNT pathway plays a pathogenic role in diabetic retinopathy and AMD [16, 17]. Accumulating evidence has demonstrated that oxidative stress and lipid peroxidation are causative factors of retinal diseases including diabetic retinopathy and AMD [8–10]. Leakage and subsequent oxidation of plasma lipoproteins in the retina may be implicated [26]. These observations prompted us to hypothesise that oxidative stress is a major cause of the WNT pathway activation in these disease conditions. The present study demonstrates that oxidative stress is responsible, at least in part, for the WNT pathway activation in retina of diabetic rats. Our results also show that several oxidants, including lipid peroxidation products, activate the WNT pathway in cultured retinal cells and endothelial cells. Mechanism studies indicate that these oxidants activate the canonical WNT pathway via stabilisation of LRP6 and increasing of LRP6 levels. These results for the first time elucidate the mechanism for WNT pathway activation in diabetic retinopathy and AMD.
The WNT signalling pathway participates in angiogenesis during embryogenesis and in pathological processes involving chronic inflammation and the development of tumours [29]. Recent evidence has linked the WNT pathway to vascular growth in murine and human retina [16]. Previously, we have shown that the WNT pathway is activated in retina from human patients with diabetic retinopathy and in animal models including streptozotocin-induced diabetic rats, Akita mice and oxygen-induced retinopathy mice [16]. The WNT pathway is also activated in VLDL receptor knockout mice, a genetic model of AMD [30]. In addition, activation of the WNT pathway alone in retinal pigment epithelial cells and in retina of normal rats upregulated angiogenic and inflammatory factors, including vascular endothelial growth factor, TNF-α, intercellular adhesion molecule 1 and nuclear factor kappa B [17]. A WNT signalling inhibitor ameliorated inflammation and neovascularisation in animal models of oxygen-induced retinopathy and AMD [16, 30]. These results, together with the incipient investigation, established the causative role of the WNT pathway in retinal neovascularisation and inflammation in diabetic retinopathy and AMD. Although compelling evidence revealed the role of oxidative stress and lipid peroxidation products in retinal diseases [8–10], no further studies were conducted to investigate the relationship between oxidative stress and WNT pathway activation in the retina. Here, we unmasked the intrinsic cause of WNT activation under these pathological conditions. As shown here, hydrogen peroxide, one of the initiators of lipid peroxidation, and different lipid peroxidation products (7-ketocholesterol, HNE, HOG-LDL) all activated the WNT pathway. Moreover, the anti-oxidant NAC blocked WNT pathway activation. These results established the causative role of oxidative stress in WNT pathway activation in diabetic retinopathy and AMD.
Oxidative stress is known to cause damage to nucleic acid, proteins and lipids [2]. One of the prevalent aldehydes generated is HNE, which is highly reactive and considered an indicator of oxidative stress and messenger in cell signalling. Due to chemical modification, HNE is able to invoke different cell signalling pathways depending on its concentration. At low and non-cytotoxic concentrations, HNE regulates cell proliferation and signal transduction, whereas at high concentrations it induces differentiation and apoptosis [31]. It is well accepted that HNE participates in the cell signalling that regulates inflammation, which represents the main driving force in many chronic human diseases. A large volume of data has clearly proved the involvement of HNE in human pathological conditions [3]. For instance, HNE-derived protein modifications are involved in lipofuscinogenesis and contribute to cell damaging effects of lipofuscin in retinal diseases such as AMD [32]. Moreover, concentrations of HNE in the plasma were significantly elevated in patients with diabetic retinopathy in comparison to diabetic patients without retinopathy [9]. Therefore, we chose HNE as a representative lipid peroxidation product for this study.
H2O2 is a reactive oxygen species as well as an initiator of lipid peroxidation [1], and can alone cause a significant increase of HNE adducts [33]. It is accepted that H2O2 modulates signalling pathways partly through its effects on lipid peroxidation [31]. Our results show that H2O2 induced over-activation of the canonical WNT pathway, indicating: (1) that free radicals in oxidative stress and subsequent lipid peroxidation are initial causative factors for activation of the canonical WNT pathway; and (2) the possible involvement of HNE in oxidative stress-mediated signalling. It is well accepted that oxidative stress activates forkhead box class O (FOXO) signalling; indeed, some publications have concluded that FOXO and TCF proteins compete for the limited pool of β-catenin under oxidative stress [34–37]. However, the interactions between the WNT signalling pathway and oxidative stress remain controversial. Previously, Shin et al. reported that WNT signalling was inhibited by a high concentration of H2O2 [38]. However, Funato et al. showed that a low concentration of H2O2 induced rapid stabilisation of β-catenin and activation of TCF in NIH3T3 or HEK293 cells, providing new insights into the cross-talk between redox and the WNT/β-catenin pathway [39]. With regard to HNE, it is well established that HNE interferes with many signalling pathways, but previous reports appear contradictory in some aspects. For example, HNE (5 μmol/l) was found to induce early neuronal cell death. Against this, Dozza et al. observed that HNE (10 μmol/l) provoked significant inhibition of GSK3β in human neuroblastoma IRM-32 cells [40]. The disparity may be ascribed to the concentration of oxidants and incubation time, as well as different cell types in these studies.
Conjugation with glutathione is the main pathway for HNE metabolism [3]. A deficiency of glutathione has been found in several diseases including inflammation, diabetes, cystic fibrosis, HIV, etc. NAC is the most effective and versatile antioxidant among other commonly used antioxidants, such as vitamin C, vitamin E and lipoic acid, since NAC supplies the cysteine required for glutathione synthesis and replenishment, reduces disulphide bonds in proteins and scavenges free radicals [22]. Our in vitro experiments showed that HNE-induced WNT pathway activation was attenuated by NAC treatment, mainly through blocking LRP6 phosphorylation and stabilisation induced by HNE, indicating that NAC renders more glutathione to conjugate HNE and prevents its interactions with target proteins. Additionally, NAC also blocked WNT ligand-induced WNT activation. Similarly, a previous study reported that WNTs and related signalling proteins are the major targets of glutathione action [23], strongly supporting the notion that WNT signalling pathway is redox-sensitive and that oxidative stress is a cause of WNT pathway activation. Moreover, placebo-controlled trials have shown that oral administration of NAC is beneficial in several diseases [22]. Our in vivo study presented here showed that NAC treatment inhibits WNT pathway activation in retina of diabetic rats and downregulates expression of a target gene of WNT signalling. These findings suggest that blockade of the WNT pathway by NAC may be a possible molecular mechanism underlying its beneficial effects.
Regarding the mechanism underlying HNE-induced activation of the WNT pathway, we showed that HNE increases phosphorylation of LRP6, an initial step in WNT pathway activation. However, the mechanism by which HNE increases phosphorylated LRP6 levels remains unclear. It is known that WNT ligands lead to WNT–frizzled–LRP6 complex formation and LRP6 phosphorylation [41–43]. The hydroxyl group in HNE makes it highly reactive with thiol of cysteine in proteins [3]. Moreover, some studies have suggested that proteins cross-link via bridging by HNE and subsequently become more resistant to proteolysis [44]. Proteasome itself is a known target of HNE, and HNE contributes to protein accumulation via impairment of ubiquitin and/or proteasome-dependent intracellular proteolysis [45]. Thus, one possible scenario is that HNE modifies LRP6 or proteasome and inhibits LRP6 degradation. This assumption is supported by our LRP6 stability assay, which showed that HNE inhibits LRP6 degradation rather than upregulating its production. The increased stability of LRP6 by HNE could contribute to the elevation of LRP6 protein levels by HNE. Moreover, HNE can react with nucleic acid and acts as a strong mutagen through a clastogenic action. HNE covalently binds to DNA and DNA-associated histones, which can affect the conformation of histones [3]. A documented study has reported that the ability of HNE to alter DNA×histone interactions could contribute to the vulnerability of DNA to oxidation, which has been observed in the brain of Alzheimer patients [46]. The covalent and non-covalent modifications of DNA and histone proteins, which is a well-known form of epigenetic regulation, is one of the mechanisms that directly influence chromatin structure [47]. Therefore, it is possible that epigenetic alteration may contribute to the increase of LRP6 protein content/phosphorylation by HNE. Since LRP6 phosphorylation itself involves multiple mechanisms and many proteins [48, 49], the molecular mechanism underlying the HNE-mediated LRP phosphorylation warrants further investigation.
In conclusion, our study established oxidative stress and lipid peroxidation products as activators of the canonical WNT pathway, a pathway that is pivotal in the pathogenesis of diabetic retinopathy and AMD. Furthermore, this study showed that the effect of HNE on the WNT signalling pathway could be through stabilisation of the WNT co-receptor, thus elucidating a pathogenic mechanism of HNE in chronic diseases.
Abbreviations
- AMD:
-
Age-related macular degeneration
- BRCEC:
-
Bovine retinal capillary endothelial cells
- CTGF:
-
Connective tissue growth factor
- FOXO:
-
Forkhead box class O
- GSK3β:
-
Glycogen synthase kinase 3β
- HNE:
-
4-Hydroxynonenal
- HOG-LDL:
-
Heavily oxidised glycated LDL
- LRP:
-
LDL receptor-related protein
- NAC:
-
N-Acetyl-cysteine
- TCF:
-
T cell factor
- WNT:
-
Wingless-type MMTV integration site
References
Mattson MP (2009) Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol 44:625–633
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128
Poli G, Schaur RJ, Siems WG, Leonarduzzi G (2008) 4-Hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev 28:569–631
Rodriguez IR, Alam S, Lee JW (2004) Cytotoxicity of oxidized low-density lipoprotein in cultured RPE cells is dependent on the formation of 7-ketocholesterol. Invest Ophthalmol Vis Sci 45:2830–2837
Anderson RE, Rapp LM, Wiegand RD (1984) Lipid peroxidation and retinal degeneration. Curr Eye Res 3:223–227
Nowak JZ (2006) Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep 58:353–363
Frank RN (2004) Diabetic retinopathy. N Engl J Med 350:48–58
Nowak M, Swietochowska E, Wielkoszynski T et al (2003) Changes in blood antioxidants and several lipid peroxidation products in women with age-related macular degeneration. Eur J Ophthalmol 13:281–286
Polak M, Zagorski Z (2004) Lipid peroxidation in diabetic retinopathy. Ann Univ Mariae Curie Sklodowska Med 59:434–437
Kopitz J, Holz FG, Kaemmerer E, Schutt F (2004) Lipids and lipid peroxidation products in the pathogenesis of age-related macular degeneration. Biochimie 86:825–831
Wodarz A, Nusse R (1998) Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol 14:59–88
Lad N, Cheshier S, Kalani Y (2008) Wnt signaling in retinal development and disease. Stem Cells Dev 18:7–16
Liu C, Li Y, Semenov M et al (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847
He X, Semenov M, Tamai K, Zeng X (2004) LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131:1663–1677
Si W, Kang Q, Luu HH et al (2006) CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol Cell Biol 26:2955–2964
Chen Y, Hu Y, Zhou T et al (2009) Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am J Pathol 175:2676–2685
Zhou T, Hu Y, Chen Y et al (2009) The pathogenic role of the canonical Wnt pathway in age-related macular degeneration. Invest Ophthalmol Vis Sci 51:4371–4379
Klein RL, Semler AJ, Baynes JW, Thorpe SR, Lyons TJ, Jenkins AJ (2005) Glycation does not alter LDL-induced secretion of tissue plasminogen activator and plasminogen activator inhibitor-1 from human aortic endothelial cells. Ann N Y Acad Sci 1043:379–389
Grant MB, Guay C (1991) Plasminogen activator production by human retinal endothelial cells of nondiabetic and diabetic origin. Invest Ophthalmol Vis Sci 32:53–64
Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65:391–426
Usatyuk PV, Parinandi NL, Natarajan V (2006) Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281:35554–35566
Atkuri KR, Mantovani JJ, Herzenberg LA (2007) N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7:355–359
Gong SP, Lee EJ, Lee ST et al (2008) Improved establishment of autologous stem cells derived from preantral follicle culture and oocyte parthenogenesis. Stem Cells Dev 17:695–712
Kuiper EJ, van Zijderveld R, Roestenberg P et al (2008) Connective tissue growth factor is necessary for retinal capillary basal lamina thickening in diabetic mice. J Histochem Cytochem 56:785–792
Nagai N, Klimava A, Lee WH, Izumi-Nagai K, Handa JT (2009) CTGF is increased in basal deposits and regulates matrix production through the ERK (p42/p44mapk) MAPK and the p38 MAPK signaling pathways. Invest Ophthalmol Vis Sci 50:1903–1910
Wu M, Chen Y, Wilson K et al (2008) Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Invest Ophthalmol Vis Sci 49:2679–2685
Tsai GY, Cui JZ, Syed H et al (2009) Effect of N-acetylcysteine on the early expression of inflammatory markers in the retina and plasma of diabetic rats. Clin Experiment Ophthalmol 37:223–231
Onorato JM, Thorpe SR, Baynes JW (1998) Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann N Y Acad Sci 854:277–290
Maiese K, Li F, Chong ZZ, Shang YC (2008) The Wnt signaling pathway: aging gracefully as a protectionist? Pharmacol Ther 118:58–81
Chen Y, Hu Y, Lu K, Flannery JG, Ma JX (2007) Very low density lipoprotein receptor, a negative regulator of the wnt signaling pathway and choroidal neovascularization. J Biol Chem 282:34420–34428
Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC (2003) Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol 50:319–336
Krohne TU, Kaemmerer E, Holz FG, Kopitz J (2009) Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp Eye Res 90:261–266
Choudhary S, Xiao T, Srivastava S et al (2005) Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol Appl Pharmacol 204:122–134
Manolagas SC, Almeida M (2007) Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol 21:2605–2614
Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC (2007) Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem 282:27298–27305
Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM (2008) Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem 283:9224–9230
Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC (2005) Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308:1181–1184
Shin SY, Kim CG, Jho EH et al (2004) Hydrogen peroxide negatively modulates Wnt signaling through downregulation of beta-catenin. Cancer Lett 212:225–231
Funato Y, Michiue T, Asashima M, Miki H (2006) The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol 8:501–508
Dozza B, Smith MA, Perry G, Tabaton M, Strocchi P (2004) Regulation of glycogen synthase kinase-3beta by products of lipid peroxidation in human neuroblastoma cells. J Neurochem 89:1224–1232
Nusse R (2003) Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development 130:5297–5305
Tamai K, Semenov M, Kato Y et al (2000) LDL-receptor-related proteins in Wnt signal transduction. Nature 407:530–535
Willert K, Brown JD, Danenberg E et al (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423:448–452
Friguet B, Szweda LI (1997) Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Lett 405:21–25
Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K (1999) 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J Biol Chem 274:23787–23793
Drake J, Petroze R, Castegna A et al (2004) 4-Hydroxynonenal oxidatively modifies histones: implications for Alzheimer’s disease. Neurosci Lett 356:155–158
Munoz-Najar UM, Sedivy JM (2010) Epigenetic control of aging. Antioxid Redox Signal. doi:10.1089/ars.2010.3250
Zeng X, Huang H, Tamai K et al (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135:367–375
Pan W, Choi SC, Wang H et al (2008) Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science 321:1350–1353
Acknowledgements
This study was supported by National Institutes of Health grants EY018659, EY012231, EY019309, P20RR024215, EY17313, a grant from the Oklahoma Center for the Advancement of Science and Technology (OCAST) and a research award from the ADA.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zhou, T., Zhou, K.K., Lee, K. et al. The role of lipid peroxidation products and oxidative stress in activation of the canonical wingless-type MMTV integration site (WNT) pathway in a rat model of diabetic retinopathy. Diabetologia 54, 459–468 (2011). https://doi.org/10.1007/s00125-010-1943-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1943-1