
Abstract
Aims/hypothesis
Inadequate chaperone function relative to client protein load in the endoplasmic reticulum triggers an adaptive unfolded protein response (UPR), including the integrated stress response (ISR), the latter being also activated by other types of stresses. It is well established that pancreatic beta cells, which synthesise and secrete insulin upon nutrient stimulation, are markedly affected by pathological disruption or excessive activation of the UPR. However, whether and how physiological nutrient stimulation affects the beta cell UPR has been little investigated.
Materials and methods
We compared the effects of increasing glucose concentrations and of endoplasmic reticulum Ca2+ emptying with thapsigargin on the UPR (X-box binding protein [Xbp1] mRNA splicing and XBP1/activating transcription factor [ATF] 6-target gene expression) and ISR (eukaryotic translation initiation factor 2A phosphorylation, ATF4 protein levels and target gene expression) in isolated rat islets.
Results
Thapsigargin strongly increased both UPR and ISR. In comparison, glucose moderately increased the UPR between 5 and 30 mmol/l, but exerted complex effects on the ISR as follows: (1) marked reduction between 2 and 10 mmol/l; (2) moderate increase parallel to the UPR between 10 and 30 mmol/l. These glucose effects occurred within 2 h, were mimicked by other metabolic substrates, but were independent of changes in Ca2+ influx or insulin secretion. Remarkably, attenuating the glucose stimulation of protein synthesis with a low concentration of cycloheximide prevented UPR activation but not ISR reduction by high glucose.
Conclusions/interpretation
Nutrient stimulation acutely activates rat islet UPR in a manner dependent on protein synthesis, while exerting complex effects on the ISR. These effects may contribute to nutrient-induced maintenance of the beta cell phenotype.
Similar content being viewed by others

Introduction
Folding of proteins destined for the secretory pathway in the endoplasmic reticulum (ER) critically depends on adequate production and function of ER chaperones such as immunoglobulin heavy chain binding protein (BiP; now known as heat shock 70 kDa protein 5 [glucose-regulated protein] [HSPA5]), glucose-regulated protein (GRP) 94 (now known as heat shock protein 90 kDa beta, member 1 [HSP90B1]), calnexin, calreticulin and protein disulphide isomerase [1]. When ER chaperone function decreases or ER client protein load increases (following ER Ca2+ emptying, ATP depletion, increased protein synthesis, etc.), the unfolded protein response (UPR) is activated. This transiently reduces protein synthesis, improves the cell capacity to fold proteins and degrade unfolded proteins and increases cellular defences against various types of stresses [2–4]. This response, which is controlled by the ER stress sensors inositol requiring (IRE) 1, activating transcription factor (ATF) 6 and pancreatic ER kinase (PERK; now known as EIF2A kinase 3 [EIF2AK3]), consists of two branches. The first branch results from the unconventional splicing of X-box binding protein 1 (Xbp1) pre-mRNA by IRE1, with a subsequent increase in active XBP1, and from the proteolytic activation of ATF6. Both transcription factors contribute to increase the production of ER chaperones such as BiP and GRP94, as well as of components of the ER-associated degradation pathway like ER degradation-enhancer mannosidase alpha-like 1 (EDEM) [5]. The second branch results from phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2α; now known as EIF2A) on Ser51 by PERK, followed by a general translational pause with selective increase in Atf4 mRNA translation and subsequent stimulation of expression of ATF4-target genes like Atf3, growth arrest and DNA damage (Gadd)153 (now known as DNA-damage-inducible transcript 3 [Ddit3], also known as Chop), Gadd34 (now known as protein phosphatase 1, regulatory [inhibitor] subunit 15A [Ppp1r15a]) and BiP [6]. Because eIF2α-Ser51 can also be phosphorylated by other kinases (EIF2AK1, 2 and 4) independently of UPR activation, events downstream from eIF2α-Ser51 phosphorylation (the shared branch of the UPR as defined by Ma et al. [7]) will be referred to here as the integrated stress response (ISR) regardless of the kinase involved [7–9].
Pancreatic beta cells, which synthesise and secrete insulin in a manner dependent on glucose concentration [10], produce high levels of several UPR and ISR components. Surprisingly, both genetic disruption and excessive activation of the UPR and ISR induce beta cell apoptosis and type 1 diabetes [11–14] (recently reviewed in [4]), suggesting that survival and function of beta cells critically depend on a continuous physiological activation of the UPR to cope with their high rate of proinsulin synthesis. More recently, it was also demonstrated that prolonged exposure to palmitate or high glucose concentrations triggers an ER stress response in insulin-secreting INS1 cells [15–17], suggesting that ER stress may play a role in beta cell gluco-lipotoxicity in type 2 diabetes.
Glucose is the main physiological regulator of protein synthesis in beta cells. As such, it can be expected to activate the UPR and ISR to compensate for an increase in ER client protein load. In support of that hypothesis, it was recently demonstrated that glucose acutely induces IRE1α phosphorylation and UPR activation in isolated mouse islets [18]. However, these effects were not accompanied by Xbp1 mRNA splicing, a hallmark of UPR activation [19]. In contrast, it has recently been shown that glucose rapidly decreases eIF2α phosphorylation and the ISR in MIN6 cells [20, 21]. To investigate whether and to what extent physiological nutrient stimulation affects the UPR in normal beta cells, we tested the effects of increasing glucose concentrations and of other metabolic substrates on the UPR and ISR in cultured rat islets, and compared these effects to those of the specific inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases, thapsigargin, which is a potent inducer of ER stress in many cell types [3].
Materials and methods
Materials
Thapsigargin, cycloheximide, succinic acid monomethyl ester (SAM), α-ketoisocaproate (KIC), l-glutamine and diazoxide were purchased from Sigma (St Louis, MO, USA). l-Leucine was from Calbiochem (San Diego, CA, USA).
Antibodies
Rabbit polyclonal anti-GADD153 (sc-575), anti-ATF4 (sc-200) and anti-XBP1 (sc-7160) antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Mouse anti-BiP antibody (clone 40) was from BD Biosciences (San Diego, CA, USA). Rabbit polyclonal anti-ACTIN antibody was from Sigma. Mouse monoclonal anti-total-eIF2α antibody was provided by D. Scheuner (Howard Hughes Medical Institute, University of Michigan, MI, USA). Rabbit anti-phospho-eIF2α antibodies were generated as described previously [20].
Islet isolation and culture
Islets were isolated from the pancreas of ∼200 g male Wistar rats (local animal facility), as described previously [22]. They were precultured for 1 week at 37°C, in the presence of 5% CO2, in serum-free RPMI-1640 medium (Gibco, Grand Island, NY, USA) containing 10 mmol/l glucose (G10), 5 g/l BSA fraction V (Roche, Basel, Switzerland), 100 IU/ml penicillin and 100 μg/ml streptomycin. Large islets (diameter >150 μm) were discarded. After preculture, islets were further cultured in medium containing 2–30 mmol/l glucose (G2, G5, G10 or G30) and various test substances, then collected for further analysis. All experiments were conducted in accord with accepted standards of humane animal care and were approved by the local Institutional Committee on Animal Experimentation.
RNA extraction and cDNA synthesis
Islet total RNA was extracted and reverse-transcribed in cDNA as described [22].
Measurement of Xbp1 mRNA splicing
cDNAs corresponding to the unspliced and spliced forms of Xbp1 mRNA (NM_001004210) were amplified simultaneously with TATA-box binding protein (Tbp) cDNA by duplex radioactive PCR [19, 22] (see Electronic supplementary material [ESM] Table 1). The spliced:total Xbp1 mRNA ratio was calculated as an indicator of Xbp1 mRNA splicing.
Real-time fluorescent PCR
Real-time PCR was performed as described elsewhere [23] (ESM Table 2). Within each PCR, sample threshold cycles were compared with a standard curve prepared by serial fourfold dilutions of an appropriate control cDNA. Relative changes in gene mRNA levels were expressed relative to the mRNA levels in control islets and normalised for relative changes in Tbp mRNA levels.
Western blotting
After rinsing with ice-cold PBS, islets were precipitated with trichloracetic acid and their proteins were solubilised in Laemmli buffer. For eIF2α phosphorylation measurements, islet protein extracts were prepared as described previously [20]. Proteins were separated by 8–12% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. The membrane was blocked (5% non-fat dry milk in Tris-buffered saline [pH 7.4] containing 0.05% Tween-20), incubated overnight with the primary antibody, washed, incubated for 1 h with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody and washed before detection of bound antibodies by enhanced chemiluminescence. Band intensities were quantified by scanning densitometry and normalised to Rouge Ponceau staining intensity.
GADD153 immunohistochemistry
Islets were washed, fixed in 1% formaldehyde and embedded in paraffin. Then, GADD153 was detected on 5 μm thick sections using anti-GADD153 antibody diluted 1:800 and the procedure described previously for haem oxygenase 1 immunodetection [22].
Measurement of protein biosynthesis
Islet protein synthesis was estimated from the incorporation of l-[3,5-3H]tyrosine (Amersham Biosciences, Bucks, UK) in total islet protein extracts during culture, as previously described [24].
Insulin secretion measurements
Insulin concentrations in the culture media were determined by RIA using rat insulin as a standard [25]. The amount of insulin secreted during the whole culture period was expressed in pmol islet−1 h−1.
Data analysis
The results are presented as means ± SEM for the indicated number of experiments. In all cases, gene mRNA and protein levels were expressed relative to the level measured in the group of islets that had been maintained under the same condition as in the last 24 h of preculture (most frequently G10).
Results
Most experiments were carried out in islets precultured for 1 week in serum-free RPMI medium containing G10 and 5 g/l BSA. These islets, which have recovered from the stress of isolation [26], have a well-preserved morphology and beta cell glucose responsiveness with low level of beta cell apoptosis [27], in marked contrast with islets cultured for several days in G5 [28]. They express total Xbp1, BiP and Grp94 mRNA at much higher levels than Tbp, and Edem, Atf3, Gadd153 and Gadd34 mRNA at similar or lower levels (ESM Table 2).
Effects of thapsigargin and increasing glucose concentrations on the UPR and the ISR in isolated islets
We first tested the effects of 18 h of culture in the presence of 10−8–10−6 mol/l thapsigargin. As shown in Fig. 1, thapsigargin dose-dependently increased insulin secretion, Xbp1 mRNA splicing and mRNA levels of BiP, Grp94, Edem, Atf3, Gadd153 and Gadd34, without affecting total Xbp1 and Tbp mRNA levels. Thapsigargin also significantly increased islet XBP1, ATF4, BiP and GADD153 protein levels (Fig. 2), the latter being detected in most islet cell nuclei (Fig. 2f). The increase in Xbp1 mRNA splicing was maximal within 2 h, while Gadd153 mRNA levels steadily increased during the first 8 h of thapsigargin treatment (data not shown). Thus, emptying ER Ca2+ with thapsigargin rapidly and strongly activates both UPR and ISR in rat islet cells.

Effects of thapsigargin (TG) and increasing glucose concentrations on insulin secretion during culture (a, b); spliced : total Xbp1 mRNA ratio (c, d); and the mRNA levels of total Xbp1 (e, f), BiP (open circles), Grp94 (filled circles), Edem (open squares) (g, h), and (i, j) Atf3 (open circles), Gadd153 (closed circles) and Gadd34 (open squares) in isolated rat islets. After 1 week of preculture in G10, the islets were cultured for 18 h in G10 plus increasing thapsigargin concentrations (a, c, e, g, i) or in G2, G5, G10 and G30 (b, d, f, h, j). Gene : Tbp mRNA ratios are expressed relative to the ratio in islets cultured in G10. Data are means ± SEM for three thapsigargin and seven glucose experiments. *p ≤ 0.05 vs islets cultured in G10 (one-way ANOVA + Newman–Keuls test). # p values for comparison of G30 vs G10 mRNA levels by unpaired t test: 0.008 for BiP, 0.007 for Grp94, 0.005 for Edem, 0.036 for Atf3, 0.003 for Gadd153 and 0.007 for Gadd34. j, Please note the logarithmic scale on the y-axis

Effects of increasing glucose concentrations and thapsigargin (TG) on XBP1, GADD153, ATF4, BiP and ACTIN protein levels in cultured rat islets. After 1 week of preculture in G10, the islets were cultured for 18 h in the presence of G2, G5, G10, G30 or G10 + 1 μmol/l thapsigargin. a–c XBP1, GADD153, ATF4, BiP and ACTIN protein levels were measured by western blot as described under Materials and methods and expressed relative to the level measured in islets cultured in G10. Key: (b) XBP1, open bars; GADD153, filled bars; (c) ATF4, open bars; BiP, filled bars; ACTIN, hatched bars. d–f, GADD153 was immunodetected on sections of islets cultured for 18 h in G2 (d), G10 (e) or G10 + thapsigargin (f). Results are representative (a, d–f) or means ± SEM (b, c) for three to four experiments. *p < 0.05 vs islets cultured in G10; #p < 0.05 for the effect of G30 vs G5 (one-way ANOVA +Newman–Keuls test). b, c Please note the logarithmic scales on the y-axes
We next tested the effects of a decrease or increase in glucose concentration. As shown in Fig. 1, 18 h culture in G2–G5 vs G10 resulted in a significant decrease in insulin secretion and Xbp1 mRNA splicing, the latter being always significantly higher than zero (p < 0.0001 by one sample t test). There were no changes in total Xbp1, Grp94, Edem and Tbp, and a strong increase in Atf3, Gadd153 and Gadd34 mRNA levels. The latter effect only partly resulted from a ∼2.3-fold increase in Atf3 and Gadd153 mRNA half-life in G2 vs G10 (ESM Fig. 1). Under these conditions, XBP1 protein levels decreased in G5 vs G10 while BiP and GADD153 protein levels increased in both G5 and G2 (Fig. 2). In G2, GADD153 increased in most islet cell nuclei (Fig. 2d). Clear increases in BiP mRNA and ATF4 protein levels were only observed in G2 vs G10 (Figs. 1h and 2a,c). Thus, lowering glucose from G10 to G5 and G2 attenuates the UPR while increasing mRNA of genes involved in ISR and corresponding protein levels.
In contrast with culture in low glucose, 18 h culture in G30 vs G10 induced a significant increase in insulin secretion, Xbp1 mRNA splicing and XBP1 protein levels without changes in total Xbp1 and Tbp mRNA levels (Fig. 1b,d,f,h,j, Fig. 2a,b). High glucose also induced a small similar increase in BiP, Grp94, Edem, Atf3, Gadd153 and Gadd34 mRNA levels (Fig. 1h,j). Under these conditions, culture in G30 vs G10 significantly increased BiP, but neither ATF4 nor GADD153 protein levels (Fig. 2b,c). When the islets were cultured for 1 week in G10 or G30 immediately after isolation, the spliced : total Xbp1 mRNA ratio was also significantly increased (from 0.18 ± 0.03 in G10 to 0.24 ± 0.02 in G30; n = 7; p < 0.05 by unpaired t test). Thus, increasing glucose from G10 to G30 for 18 h up to 1 week moderately activates the UPR and increases mRNA levels of genes involved in ISR in cultured rat islets.
Altogether, these results suggest that, in contrast to thapsigargin, which strongly increases both UPR and ISR, glucose moderately increases the UPR between G5 and G30 but exerts complex effects on the ISR, namely a marked reduction from G2 to G10, and a moderate increase parallel to the UPR from G10 to G30. The effects of glucose on Xbp1 mRNA splicing and Gadd153 mRNA levels were similar in medium containing 10% FCS instead of BSA (data not shown).
Time-dependence of the effects of glucose on the UPR
We next tested the kinetics of UPR regulation by glucose between G5 and G30. After 18 h culture in G5, the rate of insulin secretion and the level of Xbp1 mRNA splicing were low. Upon stimulation with G30, the increase in insulin secretion rate and Xbp1 mRNA splicing were maximal within 2 h (Fig. 3a,b). In comparison, BiP and Grp94 mRNA levels did not increase before 4–8 h post stimulation, while Edem mRNA levels only rose after 24 h in G30 (data not shown). Conversely, in the reverse protocol, lowering glucose from G30 to G5 led to a rapid decline in insulin secretion and Xbp1 mRNA splicing (Fig. 3c,d).

Kinetics of glucose-induced changes in insulin secretion (a, c) and spliced : total Xbp1 mRNA ratio (b, d) in islets precultured for 1 week. a, b The islets were cultured for 18 h in G5 before being cultured in G5 (open symbols) or G30 (closed symbols) for 30 min up to 24 h. c, d The islets were cultured for 18 h in G30 before being cultured in G30 (closed symbols) or G5 (open symbols). The rate of insulin secretion was calculated over the whole culture period of each sample. Data are means ± SEM for three to four experiments. *p < 0.05 or less vs G5 (a, b) or G30 (c, d) (one-way ANOVA + Dunnett’s test)
Time-dependence of the effects of glucose on the ISR
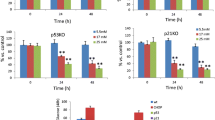
We also tested the kinetics of ISR regulation by glucose. After culture in G10, lowering glucose to G2 triggered a rapid increase in Gadd153 mRNA levels (Fig. 4a). Under these conditions, 2 h of culture in G2 increased eIF2α-Ser51 phosphorylation to a lesser extent than addition of 10−6 mol/l thapsigargin in G10, whereas culture in G5 and G30 had no effect (Fig. 4b). Interestingly, eIF2α phosphorylation was never below detection limit in the present study, even after culture in G10 (also [20]). These results confirm that lowering glucose from G10 to G2 acutely induces the ISR. To further document the opposite effects of glucose on islet UPR and ISR, we also measured Gadd153 mRNA levels and eIF2α-Ser51 phosphorylation under conditions leading to a rapid increase in Xbp1 mRNA splicing (see Fig. 3a,b). After 18 h culture in G5, stimulation with G30 led to a rapid decrease in Gadd153 mRNA levels (Fig. 4c). Under these conditions, the ratio of phosphorylated : total eIF2α was significantly reduced by 2 h culture in G30 vs G5 and increased by 2 h treatment with thapsigargin (Fig. 4d). The clear parallel between glucose-induced early changes in islet eIF2α phosphorylation and Gadd153 mRNA levels under these various conditions supports the hypothesis that culture in low glucose induces eIF2α phosphorylation and downstream activation of the ISR.

Acute effects of glucose on Gadd153 mRNA levels (a, c) and eIF2α-Ser51 phosphorylation (b, d) in islets precultured for 1 week. After preculture in G10, the islets were cultured in G10 (closed squares) or G2 (open squares) for 30 min up to 24 h (a) or for 2 h in G2, G5, G10, G30 or G10 + 1 μmol/l thapsigargin (TG) (b). c, d The islets were cultured for 18 h in G5 before being cultured in G5 (open circles) or G30 (closed circles) for 30 min up to 24 h (c) or for 2 h in G5, G30 or G5 + 1 μmol/l thapsigargin (d). Gadd153:Tbp mRNA ratios are expressed relative to the ratio measured in islets at time 0. The levels of phosphorylated (eIF2αP) and total eIF2α (eIF2αtotal) protein were measured by western blot and the ratio eIF2αP:total was expressed relative to the mean ratio in G10 (b) or G5 (d). Results are means ± SEM for three to four experiments. **p < 0.01 vs islets cultured in G10 (a, b) or G5 (c, d)(one-way ANOVA + Newman–Keuls test)
Role of mitochondrial metabolism, Ca2+ influx and insulin secretion
Glucose stimulation of beta cells increases glycolysis and mitochondrial metabolism, leading to a rise in cytosolic ATP:ADP ratio, subsequent closure of ATP-dependent K+ channels, plasma membrane depolarisation and opening of voltage-dependent Ca2+ channels. The resulting increase in cytosolic Ca2+ concentration is the triggering signal for exocytosis [10]. Except for changes in glycolysis, these stimulus–secretion coupling events can be elicited by various mitochondrial substrates in the presence of a non-stimulatory glucose concentration [29, 30]. As shown in Table 1, addition of a combination of 5 mmol/l leucine and 5 mmol/l glutamine, 10 mmol/l SAM or 5 mmol/l KIC to G5 stimulated insulin secretion and Xbp1 mRNA splicing while reducing Gadd153 mRNA expression, SAM and KIC being as effective as G10. Thus, the effects of glucose on islet UPR and ISR are reproduced by various metabolic substrates that bypass glycolysis.
To evaluate the role of plasma membrane depolarisation, stimulation of Ca2+ influx and insulin secretion, we tested the effects of diazoxide, a drug that reduces Ca2+ influx and insulin secretion by opening ATP-dependent K+ channels [10]. As expected, addition of diazoxide to G10 and G30 significantly reduced insulin secretion. However, it did not alter Xbp1 mRNA splicing or Gadd153 mRNA levels (Table 1). Similar results were obtained when insulin secretion was inhibited by clonidine, an α2-adrenergic agonist that inhibits cAMP production [31] and uncouples exocytosis from the rise in cytosolic Ca2+ concentration [32] (data not shown). Thus, glucose activates the UPR and reduces the ISR independently of its effects on beta cell membrane potential, Ca2+ influx and insulin secretion.
Role of protein synthesis
Acceleration of mitochondrial metabolism by glucose and other metabolic substrates rapidly stimulates beta cell protein synthesis with a preferential effect on proinsulin [33, 34]. To investigate the role of changes in ER client protein load in the glucose regulation of the UPR and ISR, we tested the effect of cycloheximide, an inhibitor of protein synthesis. Under control conditions, islet protein synthesis was markedly increased between G2 and G10 and slightly decreased between G10 and G30 (Fig. 5a). Cycloheximide dose-dependently inhibited protein synthesis in G10 with a half-maximal effective concentration of ∼0.1 μmol/l (data not shown). At that concentration, cycloheximide did not affect islet protein synthesis in G2, but reduced it in G5, G10 and G30 (Fig. 5a). This attenuation of the glucose stimulation of protein synthesis completely prevented the glucose stimulation of Xbp1 mRNA splicing, as well as BiP, Grp94 and Edem mRNA expression (Fig. 5c–f), but affected neither glucose-induced insulin secretion (Fig. 5b), nor glucose-induced reduction in Atf3, Gadd153 and Gadd34 mRNA levels (Fig. 5g,h; also data not shown). In contrast, cycloheximide failed to reduce the stimulation of Xbp1 mRNA splicing triggered by thapsigargin in G10, while completely preventing the increase in BiP mRNA levels (ESM Fig. 2a,b). After 1 day of culture in G5, cycloheximide also completely prevented the stimulation of Xbp1 mRNA splicing triggered by 2 h culture in G30, without counteracting the reduction in Gadd153 mRNA levels (ESM Fig. 2c,d). Thus, reducing the glucose stimulation of protein synthesis, and hence of ER protein load, abrogates the glucose stimulation of islet UPR while leaving the reduction of the ISR unaffected.

Effects of cycloheximide on glucose-induced changes in total protein synthesis (a), insulin secretion during culture (b), spliced : total Xbp1 mRNA ratio (c) and BiP, Grp94, Edem, Gadd153 and Atf3 mRNA levels (d–h) in isolated rat islets. After 1 week of preculture in G10, the islets were cultured for 18 h in G2, G5, G10 or G30 without (open circles) or with cycloheximide 0.1 μmol/l (closed circles). Gene:Tbp mRNA ratios are expressed relative to the ratio in islets cultured in G10. Data are means ± SEM for three to four experiments. *p < 0.05 for the effects of glucose vs G10 in the presence or absence of cycloheximide; # p < 0.05 for the effect of cycloheximide under the same condition (Two-way ANOVA + Bonferroni’s test). g–h Please note the logarithmic scales on the y-axes
Discussion
This study demonstrates that stimulation of isolated rat islets with glucose and other nutrients induces a rapid and sustained activation of the UPR, the maximal intensity of which is less than that induced by 10−7 mol/l thapsigargin. Thus, like thapsigargin, glucose rapidly increases Xbp1 mRNA splicing (a specific marker of IRE1 and UPR activation [19]) and XBP1 protein levels without increasing total Xbp1 mRNA levels. This effect is unrelated to the stimulation of Ca2+ influx and insulin secretion, but is likely to result from the stimulation of protein synthesis in beta cells. Thus, glucose-induced UPR activation was unaffected by diazoxide or clonidine, but was abrogated by cycloheximide at a concentration that did not suppress protein synthesis, only preventing its stimulation by glucose (Fig. 5a). Although the cycloheximide-mediated reduction in mRNA levels of UPR-responsive genes (BiP, Grp94 and Edem) may result from reduced synthesis of UPR transcription factors, the decrease in Xbp1 mRNA splicing can unambiguously be attributed to a reduction in IRE1 activity upon inhibition of protein synthesis. Thus, cycloheximide fails to inhibit Xbp1 mRNA splicing in response to thapsigargin treatment, indicating that IRE1, when adequately stimulated, is still able to splice Xbp1 pre-mRNA in cycloheximide-treated islets. Our results therefore strongly suggest that activation of the UPR by high glucose results from an increase in ER client protein load (Fig. 6b). The observation that Xbp1 mRNA splicing never reaches zero even after culture at low glucose suggests that the basal rate of protein synthesis in G2 is sufficient to slightly activate the UPR in cultured rat islets.

Nutrient regulation of the mRNA levels of genes involved in the UPR and ISR in pancreatic beta cells. a Glucose-induced changes in Xbp1 mRNA splicing (UPR, open circles) and average changes in Atf3, Gadd153 and Gadd34 mRNA levels (ISR, closed squares) relative to their level after culture in G10 (normalised data from Fig. 1d and h). b Proposed model (for explanation see Discussion). Unbroken line with arrow, stimulation; dashed line with lateral symbol, inhibition
Our results slightly contrast with a recent study showing that glucose increases IRE1α phosphorylation and expression of genes involved in UPR without increasing Xbp1 mRNA splicing in isolated mouse islets [18]. This discrepancy is unlikely to be due to species differences, because glucose also increases Xbp1 mRNA splicing in mouse islets under our culture conditions (unpublished observations, M. Bensellam). It may rather result from the use of freshly isolated vs precultured islets, since Xbp1 mRNA splicing was significantly higher in fresh vs cultured rat islets (spliced : total Xbp1 mRNA ratio 0.55 ± 0.02 in fresh islets vs 0.20 ± 0.01 in islets precultured for 1 week; n = 21–28; p < 0.0001 by unpaired t test). This high level of Xbp1 mRNA splicing, which may result from the stress of isolation, was observed regardless of the pre-isolation condition, thereby preventing us from studying the impact of in vivo hyperglycaemia on UPR activation.
What could be the physiological role of nutrient-activation of the UPR in islet beta cells? Its rapid onset upon glucose stimulation and fast reduction upon lowering of glucose concentration suggest that meal-related fluctuations in plasma nutrient concentration may induce synchronous fluctuations of Xbp1 mRNA splicing and other early steps of the UPR in beta cells. By adapting the ER chaperone to client protein ratio and the ER-associated degradation capacity to physiological variations in protein synthesis, temporal integration of these UPR fluctuations could contribute to the plasticity of the beta cell phenotype [35]. Our hypothesis may seem at variance with the observation, by electron microscopy, of normal beta cells in Xbp1 knockout mice with a liver-specific rescue of Xbp1 expression [36]. However, the argument is not sufficient to exclude a role for XBP1 in beta cell plasticity for several reasons. First, because apolipoprotein E mRNA is expressed in rat islets (unpublished observations, M. Bensellam), expression of Xbp1 under the apolipoprotein E promoter may have corrected XBP1 deficiency not only in the liver but also in beta cells. Second, beta cell function was not evaluated in that study, leaving open the possibility that beta cells were defective in those mice despite their normal morphology. Third, it is possible that XBP1 is not important in the early post-natal period, but becomes so later in life, when beta cells are more responsive to nutrient stimulation.
Although we had expected the ISR to be activated by nutrients in parallel with Xbp1 mRNA splicing, as after thapsigargin treatment, we unexpectedly observed a partial dissociation between both responses. Thus, the UPR was low in G2–G5 and concentration-dependently activated by G10 and G30, whereas the ISR was high in G2, minimal (though not zero) in G10 and slightly increased in G30, as shown by changes in eIF2α phosphorylation, ATF4 protein levels and ATF4-target gene mRNA levels. It has also been recently shown in MIN6 cells that low glucose strongly induces the ISR in beta cells [21]. As there were no detectable increases in eIF2α phosphorylation and ATF4 production in G5 vs G10, the rise in ATF4-target gene mRNA levels in G5 may have resulted from their transcriptional activation by a mechanism different from the ISR and from their concomitant stabilisation. However, our interpretation that the ISR is activated in G5 is supported by the observations that eIF2α phosphorylation rapidly decreases upon stimulation from G5 to G30 in rat islets (see Fig. 4d) and within the whole physiological range of glucose concentrations in MIN6 cells [20, 37].
It was not our primary aim to investigate the mechanism of ISR activation in low glucose. Nevertheless, our results clearly demonstrate that ISR activation can be prevented or acutely reduced by stimulation with glucose (and SAM, KIC and leucine + glutamine) independently of the stimulation of Ca2+ influx, insulin secretion and protein synthesis. It could result either from the activation of any eIF2α kinase, among which PERK and EIF2AK4 are the most likely candidates [11], or by the inactivation of protein phosphatase 1, mediated by inhibitor-1, which is highly expressed in beta cells [20]. In other cell types, complete glucose deprivation is known to induce the UPR, an effect generally attributed to ER Ca2+ emptying and reduced ER chaperone function at very low ATP concentrations [1, 3]. However, the situation was clearly different in G2–G5, where the islet ATP:ADP ratio is only moderately reduced as compared with G10 [27, 38] and Xbp1 mRNA splicing was not increased.
Although we did not fully investigate the mechanisms behind the asymmetric V-shaped glucose-concentration response curve for changes in expression of genes involved in islet ISR, our results allow us to propose the following model. On the one hand, the ISR is activated in parallel with nutrient activation of the classical UPR (pathway 1, Fig. 6b), as shown by the parallel increases in Xbp1 mRNA splicing and in mRNA levels of genes involved in UPR and ISR between G10 and G30. This ISR activation is very low, as neither eIF2α phosphorylation nor an increase in ATF4 or GADD153 protein expression could be detected in G30 vs G10. The ∼1.6-fold increase in ATF4-target gene mRNA levels in G30 vs G10 is, however, compatible with our estimation (based on changes in Xbp1 mRNA splicing) that the maximal glucose activation of the UPR is less than that produced by 10−7 mol/l thapsigargin. On the other hand, low glucose concentrations (G2 and G5 vs G10) rapidly increase mRNA levels of genes involved in ISR by inducing their transcription and, to some extent, increasing their mRNA stability (pathway 2, Fig. 6b). The reduction in mRNA of genes involved in ISR and in corresponding protein levels between G5 and G10, despite concurrent activation of the UPR, therefore indicates that, within that physiological range of glucose concentrations, pathway 2 predominates over pathway 1 for regulation of the ISR (Fig. 6b).
What could be the role of the asymmetric V-shaped glucose concentration-dependent changes in ISR in islet beta cells? In the short term, changes in eIF2α phosphorylation may play a role in the nutrient regulation of protein synthesis [12, 14, 20, 37]. This hypothesis is compatible with our finding of an inverse correlation between glucose-induced changes in mRNA levels of genes involved in islet ISR and total protein synthesis after 18 h culture. It is strongly supported by the observation that inhibiting glucose-induced eIF2α de-phosphorylation markedly reduced the acute glucose stimulation of protein synthesis in MIN6 cells [20]. In the long term, beta cell apoptosis after 2–7 days of culture also displayed a V-shaped glucose concentration-dependent curve [27, 28, 39, 40]. We, in fact, confirmed, by measuring caspase activation, that islet cell apoptosis is minimal after 1 week of culture in G10 and significantly increased by G2–G5 and, to a lesser extent, by G30 (ESM Fig. 3). The rather good correlation between rapid ISR activation and slower induction of apoptosis reinforces the hypothesis of a causal link between both events under various conditions [13, 41]. Thus, although the activation of islet ISR and apoptosis by G30 vs G10 was rather small in comparison to that induced by low glucose, the sustained activation of the islet UPR by G30 is compatible with recent studies indicating that prolonged exposure to high glucose or high NEFA concentrations induces an ER stress response that may contribute to the slow deterioration of beta cell function and survival in type 2 diabetes [15–17, 42, 43]. In this context, it will be crucial to clarify whether ISR activation contributes to beta cell apoptosis [13, 41, 44], protects against it [45, 46] or is just a marker of beta cell exposure to pro-apoptotic stimuli.
In conclusion, our results demonstrate that nutrient stimulation of rat islets moderately activates the classical UPR in a manner dependent on protein synthesis and inhibits the strongly increased expression, induced by low glucose concentrations, of genes involved in ISR. Both effects lead to an asymmetric V-shaped glucose-concentration response curve for changes in islet ISR. The kinetics of these effects suggests their possible role in nutrient-induced changes of the beta cell phenotype under both physiological and pathological conditions.
Abbreviations
- ATF:
-
activating transcription factor
- BiP:
-
immunoglobulin heavy chain binding protein
- EDEM:
-
ER-degradation enhancer mannosidase alpha-like 1
- eIF2α:
-
eukaryotic translation initiation factor 2A
- ER:
-
endoplasmic reticulum
- G10:
-
10 mmol/l glucose (G2, G5, G30 for 2, 5, 30 mmol/l glucose)
- GADD:
-
growth arrest and DNA damage
- GRP:
-
glucose regulated protein
- IRE:
-
inositol requiring
- ISR:
-
integrated stress response
- KIC:
-
α-ketoisocaproate
- SAM:
-
succinic acid monomethyl ester
- PERK:
-
pancreatic ER kinase
- UPR:
-
unfolded protein response
- XBP:
-
X-box binding protein
References
Fink AL (1999) Chaperone-mediated protein folding. Physiol Rev 79:425–449
Ma Y, Hendershot LM (2004) ER chaperone functions during normal and stress conditions. J Chem Neuroanat 28:51–65
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459
Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167:27–33
Ma Y, Hendershot LM (2004) Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem 279:13792–13799
Harding HP, Zhang Y, Zeng H et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Jiang HY, Wek SA, McGrath BC et al (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24:1365–1377
MacDonald PE, Joseph JW, Rorsman P (2005) Glucose-sensing mechanisms in pancreatic β-cells. Philos Trans R Soc Lond, B Biol Sci 360:2211–2225
Scheuner D, Song B, McEwen E et al (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Harding HP, Zeng H, Zhang Y et al (2001) Diabetes mellitus and exocrine pancreatic dysfunction in Perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell 7:1153–1163
Oyadomari S, Koizumi A, Takeda K et al (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109:525–532
Scheuner D, Vander Mierde D, Song B et al (2005) Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 11:757–764
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: role of NF-κB and endoplasmic reticulum stress. Endocrinology 145:5087–5096
Wang H, Kouri G, Wollheim CB (2005) ER stress and SREBP-1 activation are implicated in β-cell glucolipotoxicity. J Cell Sci 118:3905–3915
Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A (2006) Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology 147:3398–3407
Lipson KL, Fonseca SG, Ishigaki S et al (2006) Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 4:245–254
Calfon M, Zeng H, Urano F et al (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96
Vander Mierde D, Scheuner D, Quintens R et al (2006) Glucose activates a PP1 mediated signaling pathway to enhance overall translation in pancreatic beta cells. Endocrinology 148:609–617
Greenman IC, Gomez E, Moore CE, Herbert TP (2007) Distinct glucose-dependent stress responses revealed by translational profiling in pancreatic β-cells. J Endocrinol 192:179–187
Jonas JC, Guiot Y, Rahier J, Henquin JC (2003) Haeme-oxygenase 1 expression in rat pancreatic β-cells is stimulated by supraphysiological glucose concentrations and by cyclic AMP. Diabetologia 46:1234–1244
Elouil H, Cardozo AK, Eizirik DL, Henquin JC, Jonas JC (2005) High glucose and hydrogen peroxide increase c-Myc and haeme-oxygenase 1 mRNA levels in rat pancreatic islets without activating NFκB. Diabetologia 48:496–505
Garcia-Barrado MJ, Ravier MA, Rolland JF, Gilon P, Nenquin M, Henquin JC (2001) Inhibition of protein synthesis sequentially impairs distinct steps of stimulus-secretion coupling in pancreatic β cells. Endocrinology 142:299–307
Heding LG (1972) Determination of total serum insulin (IRI) in insulin-treated diabetic patients. Diabetologia 8:260–266
Jonas JC, Laybutt DR, Steil GM et al (2001) High glucose stimulates early response gene c-Myc expression in rat pancreatic β cells. J Biol Chem 276:35375–35381
Khaldi MZ, Guiot Y, Gilon P, Henquin JC, Jonas JC (2004) Increased glucose sensitivity of both triggering and amplifying pathways of insulin secretion in rat islets cultured for one week in high glucose. Am J Physiol, Endocrinol Metab 287:E207–E217
Van de Casteele M, Kefas BA, Cai Y et al (2003) Prolonged culture in low glucose induces apoptosis of rat pancreatic β-cells through induction of c-myc. Biochem Biophys Res Commun 312:937–944
Malaisse WJ, Sener A, Malaisse-Lagae F et al (1982) The stimulus-secretion coupling of amino acid-induced insulin release. Metabolic response of pancreatic islets of L-glutamine and L-leucine. J Biol Chem 257:8731–8737
Malaisse WJ, Rasschaert J, Villanueva-Penacarrillo ML, Valverde I (1993) Respiratory, ionic, and functional effects of succinate esters in pancreatic islets. Am J Physiol 264:E428–E433
Schuit FC, Pipeleers DG (1986) Differences in adrenergic recognition by pancreatic A and B cells. Science 232:875–877
Renstrom E, Ding WG, Bokvist K, Rorsman P (1996) Neurotransmitter-induced inhibition of exocytosis in insulin-secreting β cells by activation of calcineurin. Neuron 17:513–522
Ashcroft SJ, Bunce J, Lowry M, Hansen SE, Hedeskov CJ (1978) The effect of sugars on (pro)insulin biosynthesis. Biochem J 174:517–526
Schuit FC, In’t Veld PA, Pipeleers DG (1988) Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci USA 85:3865–3869
Hinke SA, Hellemans K, Schuit FC (2004) Plasticity of the β cell insulin secretory competence: preparing the pancreatic β cell for the next meal. J Physiol 558:369–380
Lee AH, Chu GC, Iwakoshi NN, Glimcher LH (2005) XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 24:4368–4380
Gomez E, Powell ML, Greenman IC, Herbert TP (2004) Glucose-stimulated protein synthesis in pancreatic β-cells parallels an increase in the availability of the translational ternary complex (eIF2-GTP.Met-tRNAi) and the dephosphorylation of eIF2α. J Biol Chem 279:53937–53946
Detimary P, Jonas JC, Henquin JC (1995) Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J Clin Invest 96:1738–1745
Hoorens A, Van de Casteele M, Klöppel G, Pipeleers DG (1996) Glucose promotes survival of rat pancreatic β cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98:1568–1574
Efanova IB, Zaitsev SV, Zhivotovsky B et al (1998) Glucose and tolbutamide induce apoptosis in pancreatic β-cells—a process dependent on intracellular Ca2+ concentration. J Biol Chem 273:33501–33507
Cardozo AK, Ortis F, Storling J et al (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes 54:452–461
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–S107
Laybutt DR, Preston AM, Akerfeldt MC et al (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50:752–763
Hartman MG, Lu D, Kim ML et al (2004) Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol Cell Biol 24:5721–5732
Boyce M, Bryant KF, Jousse C et al (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307:935–939
Yusta B, Baggio LL, Estall JL et al (2006) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 4:391–406
Acknowledgements
The authors thank A. Dannau and F. Knockaert for expert technical help and D. Scheuner (University of Michigan, Ann Arbor, MI, USA) for supply of the eIF2α antiserum. This work was made possible by a grant from the European Foundation for the Study of Diabetes/MSD Research Partnership for European Studies on beta cell function and survival. It was supported by Grant 1.5.182.04 from the Fonds de la Recherche Scientifique (Scientific Research Foundation; FRS-FNRS, Belgium), Grants 3.4616.05 and 3.4506.05 from the Fonds de la Recherche Scientifique Médicale (Medical Research Foundation, Belgium), the Interuniversity Poles of Attraction Program (P5/17)-Belgian Science Policy, Grant ARC 05/10-328 from the General Direction of Scientific Research of the French Community of Belgium and Grant (GOA/2004/11) from the Catholic University of Leuven (to F. C. Schuit). M. Bensellam is Research Fellow and J. C. Jonas is Senior Research Associate of the FRS-FNRS (Belgium).
Duality of interest
The authors have no duality of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Elouil, H., Bensellam, M., Guiot, Y. et al. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 50, 1442–1452 (2007). https://doi.org/10.1007/s00125-007-0674-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0674-4