
Abstract
We review the progress in testing the thrifty phenotype hypothesis. Many human epidemiological studies both by ourselves and others have confirmed and extended the original observations on which the hypothesis was based. We are not aware of any contradictory findings and we emphasise the strength of the association between birth weight and the subsequent development of the metabolic syndrome. We have worked extensively experimentally to test the hypothesis in a rat model in which pregnant and/or lactating dams are fed a diet moderately restricted in proteins. The range of programming effects that we have discovered in this example of fetal and early postnatal growth restriction is listed and includes changes in hormone receptors, signalling molecules and regulatory enzymes. We have shown the model to develop diabetes, the metabolic syndrome and signs of premature renal failure. We summarise these and other similarities between the phenotype of this model and human Type 2 diabetes and the metabolic syndrome. The number of insults during early development which can lead to a similar outcome is discussed and the suggestion is made that the early life response to stress is limited in its flexibility with outcomes including ageing and decreased longevity. Our preliminary results indicate that some MODY genes could suggest pathways whereby the changes occur and that epigenetic changes during development are involved. We conclude that the way is now clear to discover early human markers of programming by early life growth restriction and to use these to devise strategies for the prevention of Type 2 diabetes.
Similar content being viewed by others


The recent sequencing of the human genome has induced many to believe that the way is now clear towards understanding the pathogenesis of most major diseases. This belief is reminiscent of a question after a lecture given by D. Hodgkin on the crystal structure of insulin: "where does glucose bind?" Knowing the structure of insulin, a major advance at the time, was only a step on the way to understanding how it functions (which we still do not fully understand even now). Equally, knowing the sequence of the human genome, whilst a major feat of modern research, is only the beginning towards understanding how the environment determines how, when, where and to what level the genome is expressed. We propose that answering these questions is the most important key to understanding the pathogenesis of Type 2 diabetes, the metabolic syndrome and some cases of renal failure.
During the 11 years since the proposal of the "thrifty phenotype" hypothesis [1] we have been systematically testing the concepts and mechanisms proposed using a rat animal model in which fetal and/or early postnatal growth restriction is produced by reducing the protein content of the maternal diet. The effects of this manipulation are then studied in the offspring in the young mature adult (3 months) and then on into old age. All offspring are weaned onto a normal diet fed ad lib except in a few instances as described below.
We briefly review more recent human epidemiological data which has emerged since the proposal of the thrifty phenotype hypothesis and the outcome of our animal experiments. From this data we propose some questions and conclusions which we believe to be appropriate to the present state of our knowledge.
Human epidemiology
Our initial studies linked early human growth restriction, evidenced by the measurement of birth weight or thinness at birth, with an increased risk of the development as an adult of poor glucose tolerance [2], insulin resistance [3] and the metabolic syndrome [4]. These effects were strong and greatly enhanced by the presence of adult obesity. Subsequently we have identified over 30 studies worldwide which have confirmed different aspects of this research [5]. We are not aware of any research which contradicts these findings, including that reported from the Leningrad study [6] which we have discussed elsewhere [7].
Other human studies have strengthened our conviction that these findings are linked to environmental rather than genetic influences.
Studies of twins [8, 9] have shown that non-genetic factors are involved. Our own twin study did not detect an effect of birth weight in twins possibly because the small total population of twins, rather than twins with poor glucose tolerance, did not provide the necessary power to reveal effects [10]. It is also possible that birth-weight reduction due to twinning involves a different process from that associated with birth-weight reduction in the general population of singletons.
Direct evidence that poor maternal nutrition can have detrimental consequences for adult glucose tolerance came from a study of adults who were in utero during the Dutch Famine towards the end of World War II. Offspring of these pregnancies were found to have highly reduced glucose tolerance, an effect most marked in those who were in utero in the last trimester of pregnancy [11].
More recently, evidence for adverse consequences of a poor maternal environment has come from studies with the outcome for glucose tolerance in offspring of mothers who smoked during pregnancy. Smoking in pregnancy, long recognised as a cause of reduced birth weight, increased the amount of Type 2 diabetes in the offspring [12].
Animal models
We have worked extensively on a rat model in which pregnant and/or lactating dams are fed an isocaloric diet containing just under half the normal protein content. This is a modest dietary insult as shown by normal rates of conception and litter size of the affected dams. Birth weight is reduced by approximately 15%. In the great majority of studies offspring have been weaned onto a normal diet fed ad lib. In a few experiments the restriction of protein was carried on post weaning up to 70 days and some animals were given a highly palatable diet to induce obesity. We chose the model not because we believe that a low-protein diet is a major factor in the cause of human Type 2 diabetes (although it may well be important in some situations), but because of prior research showing its detrimental effects on offspring beta cells and islet vascularisation [13]. We therefore chose to focus most of our work on insulin's action in vivo and in vitro on isolated liver, muscle and adipose tissue.
Studies in vivo
Glucose tolerance, studied by the intraperitoneal injection of glucose in unanesthetized animals, showed clear age-dependent changes in the offspring of low-protein pregnancies.
In early life (6 weeks–3 months) glucose tolerance was increased and plasma insulin concentrations reduced, indicating an increased insulin sensitivity [14, 15, 16]. By the age of 1 year this effect had disappeared and there was no difference in glucose tolerance. However at 15 months there was impaired glucose tolerance [16] and by 17 months in males the emergence of frank diabetes [17]. Insulin concentrations in males at 17 months were approximately doubled, clearly indicating insulin resistance at this stage [17].
Studies of blood pressure in the offspring of low-protein pregnancies have shown it to be strongly influenced by the post-weaning diet [18, 19, 20]. In our studies of more prolonged low-protein exposure, coupled to the use of diet-induced obesity, both early growth restriction and obesity individually caused raised blood pressure. The two influences when added together caused a still further and very substantial increase in blood pressure [20]. Animals subjected to early growth restriction and obesity showed all the main features of the human metabolic syndrome being short, fat, glucose-intolerant, insulin-resistant and hypertriglyceridaemic. However only in the effects on blood pressure were the effect of the two influences found to be additive.
Plasma catecholamines (adrenalin and nor-adrenalin) in the fed state were increased in the offspring of low-protein pregnancies. Fasting, whilst producing a rise in control offspring, produced no further rise in the low-protein offspring [21]. The latter also had increased concentrations of NEFA in their plasma. These changes, coupled with changes in receptors, we believe indicate a change in the metabolic setting of the low-protein offspring such that their metabolism even when fed is more similar to that of the fasting state. This we suggest is part of the changed set point of metabolism in the thrifty phenotype which could aid survival in animals exposed to intermittent and poor nutrition.
Studies in vitro
Although a focus of our research has been, for obvious reasons, on endocrine control, we have nevertheless been impressed with the frequency with which we have found long-term (programmed) changes in the expression of hormone receptors. These include those for insulin and catecholamines (increased) and glucagon (decreased). This is perhaps not surprising if the biological advantages of an adaptation to poor nutrition are that the responses to it are such that they reduce the detrimental effects of that environment. These responses in turn will very much revolve around the effects of hormones.
Increased expression of the insulin receptor [22, 23, 24] has been observed and this reversed with age [25] possibly providing part of the explanation of the change from increased to decreased insulin sensitivity with age.
In contrast to the increased insulin-stimulated glucose uptake in both muscle and adipose tissue of 3-month-old offspring is the reduction of insulin inhibition of lipolysis in adipose tissue [26]. Coupled with the increased sensitivity of lipolysis to catecholamines (due at least in part to increased expression of their receptors) this could explain raised plasma NEFA concentrations in offspring in the fed state [15].
Striking changes in the expression of the insulin-signalling enzyme, phosphatidylinositol 3-kinase could underlie selective alterations in the down-stream insulin-regulated events. The beta form of the catalytic subunit of this enzyme was drastically reduced [24].
The changes summarised above in the in vitro properties of tissues taken from low-protein offspring could of course represent the enduring effects in vitro of changes in hormone concentrations to which the tissues were exposed in vivo. We believe that this is unlikely to be the explanation as the studies completed thus far have shown the persistence of changes despite the isolation and multiplication of cells in tissue culture. Thus when pre-adipocytes from 15-month low-protein offspring were allowed to replicate in vitro to produce an approximately tenfold increase in cell number, the increase of basal glucose uptake and resistance to the action of insulin to stimulate glucose uptake persisted [27].
The inevitable conclusion from these experiments is that environmental factors experienced in early life can result in changes in gene expression i.e. epigenetic effects. A consequence of this conclusion is that knowledge of a genotype alone does not allow the deduction of the phenotype that it will give rise to. It is essential to know how environmental events have modified gene expression before the phenotype can be deduced.
The range of programmed molecular changes which we have observed so far are summarised in Table 1.
From these changes we can deduce certain advantageous features (as judged from the likelihood of survival to successful reproduction). These can be summarised as:
- i):
-
Increased insulin sensitivity of muscle and adipose tissue. This would allow for the avid uptake and storage of nutrients such as glucose and the synthesis of macromolecules on the occasions when food is available.
- ii):
-
Raised catecholamines and NEFA in the fed state, increased adipose tissue catecholamine sensitivity and a decreased effect of insulin to inhibit lipolysis. These changes will bias energy metabolism to fat oxidation with conservation of glucose.
- iii):
-
Reduced hepatic glucagon sensitivity. This will lead to a reduced rate of ketone body production at a given concentration of circulating NEFA. In this way animals sustain raised NEFA concentration with a reduced risk of ketoacidosis [15].
Of note, in proposing the "thrifty genotype" hypothesis Neel speculated that this might include a massive secretion of insulin during the (rare) times of nutrient consumption, thereby ensuring efficient retention and storage of nutrients [30]. Our model of the thrifty phenotype suggests that this outcome could be more efficiently, effectively and selectively attained by increases in the expression of the insulin receptor in specific tissues. It is also interesting to consider whether the process by which the conditions experienced by the mother appear to be communicated to, and result in an appropriate adaptive response by the fetus, is limited to nutrition or whether it could be a more general physiological principle. In this context it is worth noting that cold conditions experienced by the mother lead to adaptive changes in the mechanisms for thermogenesis by the newborn [31]. There could therefore be a more general regulatory phenomenon shown by these studies.
The studies summarised above show conclusively that programming of metabolism can be established during fetal life. However, it is only when this (normal) phenomenon clashes with the actual postnatal environment that disease results. For example, in rural Sub-Saharan Africa where poor nutrition, high rates of physical activity and fetal growth restriction exist, obesity and diabetes are extremely rare. Conversely in populations moving rapidly from poor nutrition in a rural environment, with low birth weights, to increased nutrition and low physical activity in an urban environment, with consequent obesity, there is an "epidemic" of diabetes.
How many insults, how many pathways?
If a low-protein diet consumed by a mother can induce the changes described, do other maternal nutritional deficiencies induce the same or a different spectrum of effects? Do other "stresses" during pregnancy, e.g. infection, physical or mental stress, lead to permanent changes in the development of the offspring and if so, do they produce a similar or different phenotype?
It is only very recently that data is beginning to emerge relevant to these important questions [32]. We have looked at another nutritional deficiency that is common during human pregnancy, namely iron deficiency [33, 34, 35]. Summarising the limited data available to date, one could tentatively conclude that the phenotypic results of stressed pregnancies are relatively similar at least with respect to glucose tolerance and blood pressure. Undoubtedly much further research remains to be done but it could be that the fetal response to such stresses is fairly limited in its flexibility.
Another important question which arises concerning fetal growth and adult illness is whether, leaving aside the question of susceptibility to individual diseases, restricted early growth affects overall longevity and the closely related phenomenon of ageing. It is important to recognise that a preoccupation with the detrimental effects of ageing is particularly, if not uniquely, human. A rationalisation of the biological benefits to be derived from the adaptations we are describing is that the key "need" for survival is to carry out successful reproduction. Very few animals in the wild survive to old age. Thus a strategy which benefits short-term survival at the expense of longevity is likely to be beneficial in the wild.
Longevity
A variant of the low-protein model during pregnancy and lactation which we have used is to limit the dietary exposure to pregnancy or lactation alone. We achieve this separation by producing simultaneous control and low-protein pregnancies and then swapping the mothers of the respective litters. Thus control mothers suckle low-protein offspring and vice versa. Whilst we still have a relatively limited set of data on this variant of the model some striking findings have emerged in relation to longevity. Male offspring which have experienced a low-protein pregnancy but which have been suckled by a normally-fed dam exhibit "catch up" growth but die young [16]. The opposite occurs with male offspring experiencing a normal pregnancy but being suckled by a protein-restricted dam. These animals live longer than the control animals [16]. We have repeated these findings and exaggerated them by manipulating the litter size [36]. Whilst our findings relate to male animals it seems that a similar trend can occur in females although the differences are much smaller [16].
The increased longevity of the low-protein suckled male rats could be related to reduced food consumption [37] since this has been widely recognised for many years' as a means of increasing longevity in animals.
It is not widely recognised that appetite can be programmed during lactation [38]. High nutrition at this time permanently increases appetite and low nutrition conversely. We suspect that this explains the increased longevity in our male rats, poorly nourished whilst suckling. We have also shown that the effect of poor nutrition during lactation is so powerful in its effect to decrease appetite that it over-rides the obesity-inducing effect of a highly palatable diet in mice [39].
It will be most important to establish whether a similar programming of appetite occurs in humans since it offers the potential for the drug-free voluntary prevention of human obesity, now a major factor in diabetes causation.
Programming of appetite during lactation provides a clear indication that environmentally controlled plasticity of development extends into the postnatal period. The extent and duration of this postnatal plasticity is important because of the opportunities it offers for intervention. For example, the postnatal repopulation of islet beta cells [40] and the postnatal establishment of the zonation of metabolism of the human liver [41] suggest that there could be considerable scope for beneficial manipulations of these processes postnatally. To intervene either antenatally or postnatally it is necessary first to understand the mechanisms which underlie these processes.
What are the mechanisms?
In putting forward the thrifty phenotype hypothesis postulating the importance of fetal developmental processes in the aetiology of Type 2 diabetes we wrote: "in thinking of candidate genes in Type 2 diabetes we should widen our horizons considerably and consider genes involved in fetal growth and development" [1].
Since then it has been established that over 70% of the genetic changes leading to MODY are in genes which code for proteins involved in the regulation of developmental processes. Observing this we considered that these "experiments of nature" might provide clues as to the proteins involved in environmentally-induced changes of development which lead to an enhanced risk of Type 2 diabetes in humans. Since the most common of these is the nuclear transcription factor HNF-1α we therefore carried out a study on this. In 3-month-old male offspring of rat dams consuming a low-protein diet during pregnancy and lactation (which were weaned onto a normal diet) HNF-1α DNA binding activity was reduced both in the liver and kidney. The expression of downstream targets regulated by this nuclear factor were also reduced in the liver and kidney [42]. We concluded that reduced HNF-1α activity could in part explain the link between low birth weight and Type 2 diabetes.
By what mechanisms does early growth alteration impact on life expectancy?
An organism endowed with a reduced number of cells in a given tissue at birth, if it succeeds in "catching up" in organ weight postnatally, must either indulge in increased postnatal cell division or increase the size of individual cells or both. Either of these processes could have detrimental long-term consequences. We therefore raised the question as to whether catch-up growth could be associated with detrimental long-term, consequences [16]. Subsequently epidemiological studies from Finland have indicated that this could also be true in the human situation both in relation to Type 2 diabetes and to coronary artery disease [43, 44].
Because of the now well-established link of telomere shortening to replication-induced cell senescence in vitro [45], we have carried out studies to determine whether the rate of telomere shortening in key tissues could be related to our observed changes in longevity. We found that in male rats aged 13 months the rate of telomere shortening in the kidney of rats with reduced longevity was greater than that seen in male rats with increased longevity [36]. That this effect was observed in the kidneys but not in the liver or brain is consistent with the finding that male rats most commonly die of renal failure [46].
The mechanism(s) responsible for kidney telomere shortening are not yet clear. The extent of the shortening observed is too great to be explained by cell replication alone. However, recently it has been observed that the sequence GGG which is highly conserved in telomeres throughout most plants and animals [47, 48] is very susceptible to oxidative damage [49] and hence accelerated shortening of telomeres [50]. As a result we have suggested that, in addition to their function to monitor and therefore limit the number of cell divisions, telomeres also monitor the amount of oxidative damage which a cell has sustained [51]. In this way cells which have undergone extensive oxidation with the risk of chromosomal fusions would stop division and become senescent.
If an important mechanism leading to renal tubular cell senescence is oxidative damage to telomeres with a consequent accelerated rate of telomere shortening then potential therapeutic strategies could emerge. Whilst this prospect must be somewhat speculative at this stage [52] publications referring to the potentially beneficial effects of antioxidants which decrease or prevent the deterioration of renal function, are beginning to emerge [53].
Conclusions
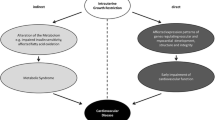
Remarkable similarities have been discovered between the phenotypic features of human Type 2 diabetes and the metabolic syndrome and the phenotype of our rat model used to test the thrifty phenotype hypothesis (Table 2).
Nevertheless many important questions remain for future research. Included in these are the questions as to how many different insults during pregnancy can lead to this phenotype and how many pathways could be involved.
It is however clear from animal experimental research that maternal diet restriction causing early growth restriction of offspring can lead to diabetes, the metabolic syndrome and renal failure. In addition much human epidemiological research suggests that fetal and early postnatal growth restriction has similar consequences in humans.
Animal research shows that the processes occurring during early life growth restriction permanently change (programme) the level of gene expression including genes involved in the production of developmental regulatory factors, hormone receptors, signalling molecules and regulatory enzymes. The inevitable conclusion from these findings is that knowledge of the structure of a particular human genome alone will not allow the accurate prediction of the resulting phenotype. Definition of the effects of environmental factors to permanently change how, when, where and to what level genes are expressed is essential for the prediction of phenotype. To obtain this information it is necessary to establish human markers which indicate the extent of environmentally-induced changes in the expression of the genome. Such markers should allow the detection of environmental change and could lead to early intervention. The ultimate hope is that such markers will lead to the establishment of intervention strategies which will allow the prevention of Type 2 diabetes and the metabolic syndrome. It is our belief that results from appropriate animal models will steer our search for these human markers.
References
Hales CN, Barker DJP (1992) Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35:595–601
Hales CN, Barker DJP, Clark PMS et al. (1991) Fetal and infant growth and impaired glucose tolerance at age 64 years. BMJ 303:1019–1022
Phillips DIW, Barker DJP, Hales CN et al. (1994) Thinness at birth and insulin resistance in adult life. Diabetologia 37:150–154
Barker DJP, Hales CN, Fall CHD et al. (1993) Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyper-lipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36:62–67
Hales CN, Barker DJP (2001) The thrifty phenotype hypothesis. Brit Med Bull 60:5–20
Stanner SA, Bulmer K, Andres C et al. (1997) Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ 315:1342–1348
Ozanne, SE, Hales, CN (1998) The thrifty phenotype hypothesis. Diabet Rev Int 7:5–7
Poulsen P, Vaag AA, Kyvik KO, Moller Jensen D, Beck-Nielsen H (1997) Low birth weight is associated with NIDDM in discordant monozygotic and dizygotic twin pairs. Diabetologia 40:439–446
Bo S, Cavallo-Perin P, Scaglione L et al. (2000) Low birthweight and metabolic abnormalities in twins with increased susceptibility to Type 2 diabetes. Diabet Med 17:365–370
Baird J, Osmond C, MacGregor A et al. (2001) Testing the fetal origins hypothesis in twins: the Birmingham twin study. Diabetologia 44:33–99
Ravelli ACJ, Meulen JHP van der, Michels et al. (1998) Glucose tolerance in adults after prenatal exposure to the Dutch famine. Lancet 351:173–177
Montgomery SM, Ekbom A (2002) Smoking during pregnancy and diabetes mellitus in a British longitudinal birth cohort. BMJ 324:26–27
Snoek A, Remacle C, Reusens B, Hoet JJ (1990) Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate 57:107–118
Shepherd PR, Crowther NJ, Desai M et al. (1997) Altered adipocyte properties in the offspring of protein malnourished rats. Br J Nutr 78:121–129
Ozanne SE, Wang CL, Petry CJ et al. (1998) Ketosis resistance in the male offspring of protein malnourished rats. Metabolism 47:1450–1454
Hales CN, Desai M, Ozanne SE et al. (1996) Fishing in the stream of diabetes: from measuring insulin to the control of fetal organogenesis. Biochem Soc Trans 24:341–350
Petry CJ, Dorling MW, Pawlak DB et al. (2001) Diabetes in old male offspring of rat dams fed a reduced protein diet. Int J Exp Diabetes Res 2:139–143
Langley-Evans SC (1996) Intrauterine programming of hypertension in the rat: nutrient interactions. Comp Biochem Physiol Physiol 114:327–333
Langley-Evans SC (2000) Critical differences between two low protein diet protocols in the programming of hypertension in the rat. Int J Food Sci Nutr 51:11–17
Petry CJ, Ozanne S E, Wang CL et al. (1997) Early protein restriction and obesity independently induced hypertension in year old rats. Clin Sci 93:147–152
Petry CJ, Dorling MW, Wang CL (2000) Catecholamine levels and receptor expression in low protein rat offspring. Diabet Med 17:848–853
Ozanne SE, Smith GD, Tikerpae J et al. (1996) Altered regulation of hepatic glucose output in the male offspring of protein malnourished rat dams. Am J Physiol 270:E55–E64
Ozanne SE, Wang CI, Coleman N et al. (1996) Altered muscle insulin sensitivity in the male offspring of protein malnourished rats. Am J Physiol 271:E1128–E1134
Ozanne SE, Nave BT, Wang CI et al. (1997) Poor fetal nutrition causes long term changes in expression of insulin signalling components in adipocytes. Am J Physiol 273:E46–E51
Ozanne SE, Dorling MW, Wang CL et al. (2001) Impaired PI 3-kinase activation in adipocytes from early growth restricted male rats. Am J Physiol (Endocrinol Metab) 280:E534–E539
Ozanne SE, Wang CI, Dorling MW et al. (1999) Dissection of the metabolic actions of insulin in adipocytes from early growth retarded male rats. J Endocrinol 162:313–319
Ozanne SE, Wang CL (2002) Adipocyte metabolism is programmed by early growth restriction. Diabetes 52 (Suppl 2):1199-P (Abstract)
Ozanne SE, Olsen GS, Hansen LL et al. (2003) Early growth restriction leads to down regulation of protein kinase C zeta and insulin resistance in skeletal muscle. J Endocrinol 177:235–241
Desai M, Byrne CD, Zhang J, Petry CL et al. (1997) Programming of hepatic insulin-sensitive enzymes in offspring of rat dams fed a protein restricted diet. Am J Physiol 272:G1083–G1090
Neel JV (1962) Diabetes mellitus: "a thrifty" genotype rendered detrimental by "progress". Am J Hum Genet 14:353–362
Symonds ME, Bryant MJ, Clarke L et al. (1992) Effect of maternal cold exposure on brown adipose tissue and thermogenesis in the neonatal lamb. J Physiol 455:487–502
Ozanne SE (2001) Metabolic programming in animals. Br Med Bull 60:143–152
Lewis RM, Petry CJ, Ozanne SE et al. (2001) Effects of maternal iron restriction in the rat on blood pressure, glucose tolerance and serum lipids in the 3 month old offspring. Metabolism 50:562–567
Lewis RM, James LA, Zhang J et al. (2001) Effects of maternal iron restriction in the rat on hypoxia-induced gene expression and fetal metabolite levels. Br J Nutr 85:193–201
Lewis RM, Forhead AJ, Petry CJ, Ozanne SE, Hales CN (2002) Long term programming of blood pressure by maternal dietary iron-restriction in the rat. Br J Nutr 88:283–290
Jennings BJ, Ozanne SE, Dorling MW et al. (1999) Early growth determines longevity in male rats and may be related to telomere shortening in the kidney. FEBS Lett 448:4–8
Ozanne SE, Jennings BJ, Hales CN (2000) Metabolic alterations following early growth retardation. In: Barker DJP (ed.) Fetal origins of cardiovascular and lung disease, pp 161–177
Oscai LB, McGarr JA (1978) Evidence that the amount of food consumed in early life fixes appetite in the rat. Am J Physiol 235:R141–R144
Ozanne SE, Lewis RL, Jennings BJ et al. (2002) Programming of appetite by early dietary manipulation. Diabet Med 19 (Suppl 2):P261 (Abstract)
Scaglia L, Cahill CJ, Finegord DT et al. (1997) Apoptosis participates in the remodelling of the endocrine pancreas in the neonatal rat. Endocrinology 138:1736–1741
Gebhardt R (1992) Metabolic zonation of the liver: regulation and implications for liver function. Pharmacol Ther 53:275–354
Moukayed M, Ozanne SE, Zhang J et al. (2002) HNF1α is a molecular link in intrauterine growth restriction induced type 2 diabetes. Diabet Med 19 (Suppl 2):1–27, A66 (Abstract)
Forsen T, Eriksson J, Tuomilehto J et al. (2000) The fetal and childhood growth of persons who develop Type 2. Diabet Ann Int Med 133:176–182
Eriksson JG, Forsen T, Tuomilehto J et al. (1999) Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 318:427–431
Lange T de (1998) Telomeres and senescence: ending the debate. Science 279:334–335
Iwasaki K, Gleiser CA, Masoro EJ et al. (1988) The influence of dietary protein source on longevity and age-related disease processes of Fischer rats. J Gerontol 43:B5–B12
Meyne J, Ratliff RL, Moyzis RK (1989) Conservation of the human telomere sequence (TTAGGG) among vertebrates. Proc Natl Acad Sci USA 86:7049–7053
Adams SP, Hartman TPV, Lim KY et al. (2001) Loss and recovery of Arabidopsis-type telomere repeat sequences 5′-(TTTAGGG)n-3′ in the evolution of a major radiation of flowering plants. Proc R Soc Lond Biol Sci 268:1541–1546
Oikawa S, Kawanishi S (1999) Site specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett 453:358–365
Zglinicki T von, Saretski G, Docke W et al. (1995) Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res 220:186–193
Jennings BJ, Ozanne SE, Hales CN (2000) Nutrition, oxidative damage, telomere shortening, and cellular senescence: individual or connected agents of aging? Mol Genet Metab 71:32–42
Hales CN (2001) Suicide of the nephron. Lancet 357:136–137
Fryer JM (2000) Vitamin E as a protective antioxidant in progressive renal failure. Nephrology 5:1–7
Brown DC, Byrne CD, Clark PMS et al. (1991) Height and Glucose tolerance in adult subjects. Diabetologia 34:531–533
Nelson RG, Tan M, Beck GJ (1999) Changing glomerular filtration with progression from impaired glucose tolerance to Type II diabetes mellitus. Diabetologia 42:90–93
Krook A, Bjornholm M, Galuska O (2002) Characterization of signal transoluction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 49:284–292
Bannerji MA, Chaiken RL, Gordon D et al. (1995) Does intra-abdominal adipose tissue in black men determine whether NIDDM is insulin-resistant or insulin sensitive? Diabetes 44:141–146
Frank JW, Saslow SB, Camilleri M et al. (1995) Mechanism of accelerated gastric emptying of liquids and hyperglycaemia in patients with type II diabetes mellitus. Gastroenterology 109:755–765
Bruce DG, Chisholm DJ, Storlien LH et al. (1992) The effects of sympathetic nervous system activation and psychological stress on glucose metabolism and blood pressure in subjects with Type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 35:835–843
Singh N (2002) Diabetes, heart rate and mortality. J Cardiovasc Pharmacol Ther 7:117–121
Maianu L, Keller SR, Garvey WT (2001) Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J Clin Endocrinol 86:5450–5456
Bjornholm M, Al-Khalili L, Dicker A et al. (2002) Insulin signal transduction and glucose transport in human adipocytes: effects of obesity and low calorie diet. Diabetologia 45:1128–1135
Acknowledgements
Our research summarised above has been supported by grants from Diabetes UK, European Union, Medical Research Council UK, National Institutes of Health USA, Parthenon Trust and Wellcome Trust. We are very grateful to A. Flack, D. Hutt and A. Wayman for their excellent technical support throughout the past 11 years.
Author information
Authors and Affiliations
Corresponding author
Additional information
*Based upon the Dorothy Hodgkin Lecture delivered by CNH at the Birmingham Meeting of Diabetes UK, March 2002
Rights and permissions
About this article
Cite this article
Hales, C.N., Ozanne, S.E. For Debate: Fetal and early postnatal growth restriction lead to diabetes, the metabolic syndrome and renal failure*. Diabetologia 46, 1013–1019 (2003). https://doi.org/10.1007/s00125-003-1131-7
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1131-7