
Abstract
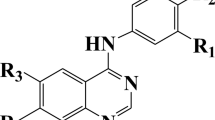
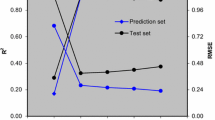
In this research, molecular docking and QSAR models based on comparative molecular field analysis-like, least squares-support vector machine, and radial basis function neural network were used to investigate the interactions of naphthyridone derivatives with the binding site of ATAD2 and predict their activities. First molecular docking was used to investigate binding interactions between molecules with the greatest, the lowest and with medium activities and the binding site of ATAD2, then comparative molecular field analysis was used to model and predict their activities. Squared correlation coefficient (R 2) for training and test sets of comparative molecular field analysis-like model and its leave-one-out cross validation (Q 2) were 0.87, 0.83, and 0.78, respectively. The contributions of steric and electrostatic fields in the building of model were 49.64 and 50.36 %, respectively. Comparative molecular field analysis contour maps were extracted and interpreted to help the design of new molecules with greater activity. Principal component analysis was performed on comparative molecular field analysis descriptors and extracted scores were used as input variable to develop more reliable least squares-support vector machine and radial basis function neural network models. R 2 values for training and test sets of least squares-support vector machine were 0.82 and 0.84, respectively, and Q 2 parameter for its training set was 0.82. These results indicate least squares-support vector machine has slightly higher predictive power compared to the comparative molecular field analysis model. R 2 values for training and test sets of radial basis function neural network model were 0.89 and 0.90, respectively, and its squared correlation coefficient for leave-one-out cross validation was 0.87 that shows radial basis function neural network model has the greatest predictive power. All models have been validated with several statistical parameters and their applicability domains show all models were reliable.




Similar content being viewed by others
References
Andersson M (2009) A comparison of nine PLS1 algorithms. J Chemometr 23:518–529
Bamborough P, Chung C-W, Furze RC, Grandi P, Michon A-M, Sheppard RJ, Barnett H, Diallo H, Dixon DP, Douault C, Jones EJ, Karamshi B, Mitchell DJ, Prinjha RK, Rau C, Watson RJ, Werner T, Demont EH (2015) Structure-based optimization of naphthyridones into potent ATAD2 bromodomain inhibitors. J Med Chem 58:6151–6178
Baroni M, Clementi S, Cruciani G, Costantino G, Riganelli D (1992) Predictive ability of regression models. Part II: Selection of the best predictive PLS model. J Chemometr 6:347–356
Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S (2013) Malignant genome reprogramming by ATAD2. BBA-Gene Regul Mech 1829:1010–1014
Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V, Barbry P, Debernardi A, Brambilla C, Brambilla E, Rousseaux S, Khochbin S (2010) Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 29:5171–5181
Chaikuad A, Petros AM, Fedorov O, Xu J, Knapp S (2014) Structure-based approaches towards identification of fragments for the low-druggability ATAD2 bromodomain. Med Chem Commun 5:1843–1848
Chung C-W, Tough DF (2012) Bromodomains: A new target class for small molecule drug discovery. Drug Discov Today Ther Strateg 9:111–120
Ciró M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, Helin K (2009) ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res 69:8491–8498
Cruciani G (2006) Molecular interaction fields: Applications in drug discovery and ADME prediction. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Dash CSK, Behera AK, Dehuri S, Cho S-B (2016) Radial basis function neural networks: a topical state-of-the-art survey. Open Comput Sci 6:33–63
Demont EH, Chung C-W, Furze RC, Grandi P, Michon A-M, Wellaway C, Barrett N, Bridges AM, Craggs PD, Diallo H, Dixon DP, Douault C, Emmons AJ, Jones EJ, Karamshi BV, Locke K, Mitchell DJ, Mouzon BH, Prinjha RK, Roberts AD, Sheppard RJ, Watson RJ, Bamborough P (2015) Fragment-based discovery of low-micromolar ATAD2 bromodomain inhibitors. J Med Chem 58:5649–5673
Filippakopoulos P, Knapp S (2012) The bromodomain interaction module. FEBS Lett 586:2692–2704
Filippakopoulos P, Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 13:337–356
Gallenkamp D, Gelato KA, Haendler B, Weinmann H (2014) Bromodomains and their pharmacological inhibitors. ChemMedChem 9:438–464
Ghasemi JB, Shiri F (2012) Molecular docking and 3D-QSAR studies of falcipain inhibitors using CoMFA, CoMSIA, and Open3DQSAR. Med Chem Res 21:2788–2806
Harner MJ, Chauder BA, Phan J, Fesik SW (2014) Fragment-based screening of the bromodomain of ATAD2. J Med Chem 57:9687–9692
Hassanzadeh Z, Ghavami R, Kompany-Zareh M (2016) Radial basis function neural networks based on the projection pursuit and principal component analysis approaches: QSAR analysis of fullerene [C60]-based HIV-1 PR inhibitors. Med Chem Res 25:19–29
Hsu K-C, Chen Y-F, Lin S-R, Yang J-M (2011) iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics 12(Suppl 1):S33
Kastenholz MA, Pastor M, Cruciani G, Haaksma EEJ, Fox T (2000) GRID/CPCA: A new computational tool to design selective ligands. J Med Chem 43:3033–3044
Malek-Khatabi A, Kompany-Zareh M, Gholami S, Bagheri S (2014) Replacement based non-linear data reduction in radial basis function networks QSAR modeling. Chemom Intell Lab 135:157–165
Ojha PK, Mitra I, Das RN, Roy K (2011) Further exploring rm2 metrics for validation of QSPR models. Chemom Intell Lab 107:194–205
Peng X, Wang Y (2009) A normal least squares support vector machine (NLS-SVM) and its learning algorithm. Neurocomputing 72:3734–3741
Richmond NJ, Willett P, Clark RD (2004) Alignment of three-dimensional molecules using an image recognition algorithm. J Mol Grap Model 23:199–209
Roy K, Kar S (2014) The rm2 metrics and regression through origin approach: Reliable and useful validation tools for predictive QSAR models (Commentary on ‘Is regression through origin useful in external validation of QSAR models?’). Eur J Pharm Sci 62:111–114
Roy K, Kar S, Das RN (2015) A primer on QSAR/QSPR modeling: Fundamental concepts, Springer international publishing.
Sahigara F, Mansouri K, Ballabio D, Mauri A, Consonni V, Todeschini R (2012) Comparison of different approaches to define the applicability domain of QSAR models. Molecules 17:4791–4810
Sanchez R, Meslamani J, Zhou M-M (2014) The bromodomain: From epigenome reader to druggable target. BBA-Gene Regul Mech 1839:676–685
Suykens JAK, Gestel TV, Brabanter JD, Moor BD, Vandewalle J (2002) Least squares support vector machines. World scientific publishing, Singapore
Tosco P, Balle T (2011) Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J Mol Model 17:201–208
Tosco P, Balle T, Shiri F (2011) Open3DALIGN: An open-source software aimed at unsupervised ligand alignment. J Comput Aided Mol Des 25:777–783
Tropsha A (2010) Best practices for QSAR model development, validation and exploitation. Mol Inform 29:476–488
Vidler LR, Brown N, Knapp S, Hoelder S (2012) Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem 2012 55:7346–7359
Wang GG, Allis CD, Chi P (2007) Chromatin remodeling and cancer, Part I: Covalent histone modifications. TRENDS Mol Med 13:363–372
Wold S, Sjöström M, Eriksson L (2001) PLS-regression: A basic tool of chemometrics. Chemom Intell Lab 58:109–130
Yun M, Wu J, Workman JL, Li B (2011) Readers of histone modifications. Cell Res 21:564–578
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Sepehri, B., Rasouli, Z., Hassanzadeh, Z. et al. Molecular docking and QSAR analysis of naphthyridone derivatives as ATAD2 bromodomain inhibitors: application of CoMFA, LS-SVM, and RBF neural network. Med Chem Res 25, 2895–2905 (2016). https://doi.org/10.1007/s00044-016-1686-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1686-8