
Abstract
Protein toxicity can be defined as all the pathological changes that ensue from accumulation, mis-localization, and/or multimerization of disease-specific proteins. Most neurodegenerative diseases manifest protein toxicity as one of their key pathogenic mechanisms, the details of which remain unclear. By systematically deconstructing the nature of toxic proteins, we aim to elucidate and illuminate some of the key mechanisms of protein toxicity from which therapeutic insights may be drawn. In this review, we focus specifically on protein toxicity from the point of view of various cellular compartments such as the nucleus and the mitochondria. We also discuss the cell-to-cell propagation of toxic disease proteins that complicates the mechanistic understanding of the disease progression as well as the spatiotemporal point at which to therapeutically intervene. Finally, we discuss selective neuronal vulnerability, which still remains largely enigmatic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The prevalence of neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Huntington’s disease (HD), is increasing at an alarming rate due to the increase in average life expectancy. Patients with these diseases display serious neurological disabilities, such as memory impairment and motor problems, for which there are no cure. One of the cardinal features of neurodegenerative diseases is the presence of protein toxicity [1]. Here, we define protein toxicity as all the pathological alterations that result from the accumulation, oligomerization, and/or multimerization of disease-associated toxic proteins.
Protein toxicity is a unifying feature of both sporadic and familial cases of neurodegenerative diseases. One of the mechanisms by which protein toxicity occurs is through genetic mutations. For example, 5 point mutations in the genes encoding synuclein alpha (SNCA; A53T, A30P, E46K, H50Q, and G51D) and 52 mutations (alzforum.org/mutations) in amyloid precursor protein (APP) have so far been identified to be associated with PD and AD, respectively [2, 3]. Likewise, protein toxicity can arise from more than 20 genetic mutations in the TARDBP gene encoding TDP-43 protein, which are associated with ALS and FTD [4]. In addition, a recently identified GGGGCC repeat expansion in the intronic region of the C9ORF72 gene is associated with ALS/FTD and is known to produce five different dipeptide-repeat proteins (DPRs; poly-GA, -GR, -GP, -PR, -PA) through repeat associated non-AUG (RAN) translation [5]. The arginine-rich DPRs, in particular, have been shown to cause protein toxicity [6]. Moreover, polyQ protein toxicity is solely caused by an expansion mutation of the glutamine tract in each of the genes responsible for polyQ diseases [7].
On the other hand, aberrant proteins generated independently of known genetic mutations can also contribute to protein toxicity. For instance, abnormal cytoplasmic accumulation of TDP-43, known as “TDP-43 pathology,” is observed in most cases of ALS and in about half of FTD cases, even when there is no TARDP mutation [8]. Likewise, independent of SNCA mutation, α-synuclein aggregation is often observed in PD and several other neurological disorders known as “synucleinopathies”. In fact, PD is rarely caused by mutations in SNCA [9]. Similarly, AD is rarely caused by mutations in APP [10], yet accumulation of amyloid beta is the hallmark feature of AD. Thus, regardless of the disease etiology (sporadic or familial), protein toxicity seems to be a hallmark of most neurodegenerative diseases.
In neurodegenerative diseases, protein toxicity in affected neurons may result in cellular defects such as transcriptional alteration, mitochondrial dysfunction, and an impaired protein/RNA quality control system, all of which critically contribute to the initiation and progression of neurodegenerative diseases. Although cell death is the final outcome of the disease process, cell death is often preceded by neurological deficits in animal models and patients [11, 12]. Hence, this review will focus on the neuronal dysfunction that occurs prior to cell death. Notably, each type of cellular defects is not absolutely specific to a certain neurodegenerative disease; but instead, these defects are more commonly observed in a variety of disease cases. Given the crucial contribution of protein toxicity to neurodegenerative disease pathogenesis, increasing our understanding of protein toxicity is indispensable for future development of rational and effective therapeutics for these diseases. Instead of characterizing protein toxicity from one disease to another (e.g., AD, PD, and ALS), in the following sections, we discuss the mechanisms underlying protein toxicity from one subcellular compartment to another (e.g., nucleus and mitochondria; see Table 1 and Fig. 1).

A schematic overview of protein toxicity. Accumulation of toxic disease proteins is shown to induce dysfunctions in specified compartments such as the nucleus, mitochondria, cytoplasm, and stress granules. They can also propagate into other nearby cells, spreading the disease pathology
Protein toxicity in the nucleus
Nuclear inclusions (NIs) of toxic proteins in neurons are observed in approximately 20 different neurodegenerative diseases [13]. In particular, nuclear accumulation of toxic disease proteins is closely associated with the pathogenesis of polyQ diseases (see below for details). Although a growing body of evidence indicates nuclear dysfunction to be central to the pathogenesis of several neurodegenerative diseases, the precise role of neuronal intranuclear inclusion bodies in the disease pathogenesis is still a matter of debate. There is a view that microscopically visible NIs are not toxic, but are instead self-protective structures or incidental byproducts of the pathogenic process. This view proposes that the more soluble protofibrillar or oligomeric aggregates (as opposed to the more mature fibrillar aggregates formed inside the nucleus) have toxic properties in afflicted neurons [14–18]. Whether or not the nuclear inclusion bodies are the major source of nuclear protein toxicity, nuclear dysfunctions such as transcriptional alteration and impaired nucleocytoplasmic transport are evident in many cases of neurodegenerative diseases [19, 20].
As described above, polyQ diseases may be representative neurodegenerative diseases associated with nuclear protein toxicity [21]. There are at least nine polyQ diseases, including HD, dentatorubral–pallidoluysian atrophy (DRPLA), spinal bulbar muscular atrophy (SBMA), and the spinocerebellar ataxias (SCAs) 1, 2, 3, 6, 7, and 17 [7]. Each of these nine polyQ diseases is caused by CAG (Q) repeat expansion mutation in each of the disease-responsible genes [e.g., the huntingtin (htt) gene mutation for HD]. Upon expansion of the Q repeats, the disease-responsible proteins, most of which mis-localize to the nucleus, gain a propensity to aggregate and multimerize with numerous target proteins. For instance, although normally a cytoplasmic de-ubiquitinase protein, ataxin-3 predominantly localizes to the nucleus upon expansion mutation in SCA3. Similarly, various animal models present nuclear aggregation of mutant htt in neurons [22]. Of note, however, the nuclear accumulation of mutated polyQ proteins is not always closely associated with the pathogenesis of diseases. In case of SCA2, it has been shown that nuclear localization of the SCA2 protein is not necessary for SCA2 pathogenesis in mice or humans [23].
PolyQ NIs are often co-localized with ubiquitin, heat shock proteins, and numerous target proteins [21]. Some proteins, such as cAMP response element-binding protein (CREB)-binding protein (CBP), have been identified as the target of polyQ proteins in a number of different polyQ diseases, suggesting that their interaction with polyQ proteins may be dependent on the expanded Q repeat region, rather than the flanking regions, of the polyQ proteins. The detection of various transcription factors, such as CBP [24–27], TATA-binding protein (TBP) [28], nuclear co-repressor (NCoR) [29], and RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) [30], within polyQ NIs suggests a sequestration mechanism by which polyQ proteins may induce transcriptional dysregulation. Notably, many of the proteins sequestered by polyQ proteins function as epigenetic regulators that may be responsible for the system-wide transcriptional dysregulation in a subset of polyQ diseases [20, 31]. Consistently, a previous study reported that the mutant htt and ataxin-3 proteins could directly bind to histone acetyl-transferases, such as CBP and p300/CBP-associated factor (P/CAF), thereby impairing histone acetylation in neurons [32, 33]. However, the reduction of histone acetylation by mutant htt remains disputed [20]. Aside from histone modification, direct epigenetic changes to the DNA have also been reported in HD. Previous studies reported hypomethylation of DNA with CpG-poor regions in the HD cell culture model [34] and a decreased level of 7-methylguanine (7mG) by mutant htt proteins in mouse and human patient samples [35]. Furthermore, a direct interaction between mutant htt proteins and methyl-CpG-binding protein 2 (MeCP2) has been reported, the interaction of which enables mutant htt to bind directly to the methylated DNA regions [36].
Transcriptional and epigenetic alterations have been shown to contribute to the broad spectrum of neuronal phenotypes ranging from early neuropathic features to late-stage neuronal cell death in polyQ diseases [31]. For instance, recent studies showed that polyQ proteins induced early changes to the dendrite morphology through the perturbation of RNA granule formation and transcriptional cascades regulating the ER-to-Golgi (COPII) pathway [37–39]. In the SCA1 mouse model, the translational repressor Capicua was shown to be critically involved [40], and in HD and DRPLA mouse models, treatment with histone-deacetyltransferase (HDAC) inhibitors (sodium butyrate, 4-phenylbutyric acid sodium salt, and suberoylanilide hydroxamic acid) was shown to ameliorate neurotoxicity [41–44]. These results demonstrate a crucial contribution of transcriptional and epigenetic alterations in at least a subset of polyQ diseases.
In addition to polyQ diseases, transcriptional dysregulation is also observed in other neurodegenerative diseases, such as AD [45–47] and PD [48, 49], although they are not generally accompanied by nuclear accumulation of toxic proteins. Similar to polyQ diseases, AD and PD also manifest epigenetic alterations, though the mechanisms of which remain to be elucidated [20, 31]. Nevertheless, treatment with certain epigenetic drugs, such as HDAC inhibitors, ameliorated AD and PD phenotypes in animal models [31], indicative of the relevance and importance of epigenetic alterations in the disease pathogenesis.
In addition to the transcriptional and epigenetic alterations, nucleocytoplasmic transport defects have emerged as one of the principal nuclear dysfunctions manifested in neurodegenerative diseases such as ALS/FTD, HD, and AD [19, 50]. The mechanisms by which nucleocytoplasmic transport becomes disrupted range from sequestration of nuclear pore complex (NPC) molecules by toxic RNA or proteins [19, 51–56] to direct blockage of nuclear pores by toxic disease proteins [57]. Some excellent reviews on this topic have recently been published, which we recommend for detailed discussion [19, 50].
Protein toxicity in the cytoplasm
Many of the disease proteins are prone to accumulate in the cytoplasm, in which the pool of potential target molecules differs significantly from that of the nucleus. For example, it is the cytoplasm in which the protein quality control (PQC) system mostly resides, not in the nucleus. The cytoplasm also contains a more elaborate cytoskeleton compared to the nucleus. Hence, due to the physical proximity, cytoplasmic protein toxicity can directly impinge on the PQC system and cargo transport via cytoskeleton disruption. In this section, we will focus on the cytoplasmic protein toxicity associated with the PQC system and the cytoskeleton.
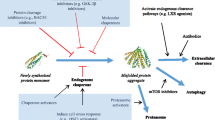
The accumulation of misfolded proteins in neurodegenerative diseases inevitably burdens the PQC system, which comprises the ubiquitin–proteasome system (UPS), chaperone-mediated autophagy (CMA), macroautophagy, and ER-associated degradation (ERAD) [58]. UPS impairment is considered to be one of the major contributing factors of neurodegenerative disease pathogenesis. Previous studies showed that aggregated beta-sheet-rich prion proteins and aggregated Tau in AD could block the 20S and 19S proteasome particles, respectively, which impaired UPS-mediated degradation [59, 60]. Consistently, genetic mutations of UPS components such as E3 ligase Parkin, deubiquitinating enzyme ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), and ATPase valosin-containing protein (VCP), can lead to neurodegeneration [61, 62]. In addition, overexpression of certain components of UPS could ameliorate the disease phenotypes in neurons in many neurodegenerative disease models [63–66]. For example, PD-associated G2019S LRRK2 mutant proteins can be ubiquitinated by E3 ligase C-terminus of HSP70-interacting protein (CHIP), whose overexpression enhances the ubiquitin proteasomal degradation of LRRK2 mutant proteins [67]. Consistently, CHIP knockout mice displayed exacerbated polyQ pathology [68]. Furthermore, mutant htt has been shown to undergo ubiquitin proteasomal degradation via E3 ligase UBE3A [69], the activity of which is down-regulated by UBR5 [70], a genetic modifier for HD [71]. Moreover, most of the protein inclusions in neurodegenerative diseases are positive for ubiquitin and chaperones, both of which become depleted in the afflicted neurons [72]. Conversely, a recent study by Nucifora and colleagues suggested that ubiquitination could be a mechanism by which protein inclusions are formed [73]. They showed that WSB1 could induce aggregation of G2019S LRRK2 via K27 and K29 ubiquitination, which appeared to be neuroprotective [73]. Ubiquitination may thus protect against protein toxicity by either inducing degradation or aggregation of toxic proteins.
CMA is a selective protein degradation system that eliminates proteins harboring a pentapeptide KFERQ-like motif, which is found in approximately 30% of cytosolic proteins [58]. When folded properly, the KFERQ motif is not exposed to the surface. However, misfolding of these proteins exposes the motif that can be subsequently recognized by the heat shock cognate protein 70 (HSC70) chaperone and CMA adaptor lysosomal membrane-associated protein 2A (LAMP-2A). Several disease-associated proteins such as LRRK2 and α-synuclein also harbor KFERQ-like motifs that are recognized by CMA for degradation [74, 75]. A previous study showed that α-synuclein proteins in PD could bind to LAMP-2A with an unusually high affinity. This strong binding in turn resulted in a “traffic jam” during cargo translocation across the lysosomal membrane, thereby inhibiting CMA [75]. As for LRRK2, its binding to the lysosomal membrane is enhanced by certain mutations, thereby facilitating accumulation of α-synuclein among other CMA substrates [74]. Moreover, PD-associated mutations in UCHL1 also interfere with the CMA process [76]. These results suggest that CMA is one of the central processes by which PD-associated proteins are degraded and that interfering with the CMA process may result in α-synuclein accumulation. In a few other studies, the augmentation of CMA was shown to enhance the removal of pathogenic disease proteins in various neurodegenerative diseases [77–79], suggesting that CMA may be an important therapeutic target for diseases associated with protein toxicity. Since aggregation-prone proteins can be efficiently eliminated by macroautophagy, its role in neurodegenerative diseases has been extensively pursued. In HD, macroautophagy activity is reduced due to the failure in cargo recognition by autophagic vacuoles [80]. In addition, a certain species of mutant htt proteins has been shown to be selective-autophagy resistant, likely due to its unconventional conformation that is unfavorable for cargo recognition by autophagic vacuoles [81]. In many neurodegenerative diseases, autophagy can be induced as a compensatory response to the failure of UPS in afflicted neurons [82–84]. However, it appears that the compensatory induction of autophagy is not enough to overcome the accumulation of ubiquitin-positive toxic proteins in HD. Consistent with this, it was shown that further genetic or pharmacological activation of autophagy has obvious therapeutic benefits in various disease models [85].
Protein toxicity commonly produces ER stress; in turn, ER stress can cause up-regulation of chaperones, ERAD and apoptotic genes, global protein translational arrest, and stress granule formation [86]. ER stress can be caused in a number of ways; one of these causes is ERAD failure. For instance, VCP, a necessary component of ERAD, was shown to be sequestered by mutant htt [87, 88]. In another study, overexpression of VCP was shown to rescue ERAD failure caused by mutant htt [89]. Interestingly, the sequestration of VCP by polyQ proteins occurs in at least four other polyQ diseases (SCA1, SCA3, SCA7, and SBMA) [90, 91], in which the loss of VCP function may be a common pathogenic mechanism.
Pathological inclusions of cytoskeletal proteins, such as neuronal intermediate filament (IF) proteins or the microtubule-associated protein tau (MAPT), are neuropathological signatures in various neurodegenerative diseases [92]. Specifically, tau-associated microtubule defects are linked to a range of neurodegenerative diseases known as “tauopathies” [93]. Changes in F-actin structures have also been reported in polyQ diseases [38] and AD [94]. Furthermore, formation of ADF/cofilin-actin filament bundles (rods) that can occlude neurites and block vesicle transport has been implicated in neurodegenerative diseases [95]. In addition to these changes in cytoskeletal structures, accumulation of toxic disease proteins can lead to defects in axonal transport [96–101]. For example, defective axonal transport was reported to be a key early feature of pathogenesis prior to neurodegeneration in various SOD1 animal models of ALS [102–106]. Various animal models of HD also showed abnormalities in both anterograde and retrograde axonal transport [107–109].
Cytoplasmic protein toxicity encompasses a whole array of neuronal phenotypes, many of which are shared among neurodegenerative diseases. Hence, therapeutically neutralizing cytoplasmic protein toxicity may be beneficial, provided that the toxic proteins remain static in the cytoplasm. However, from the cytoplasm in which toxic disease proteins are first made, these proteins can be transported to other organelles such as the nucleus (discussed above), the stress granules (discussed later), or the mitochondria (discussed next), all of which can complicate any attempts to remedy cytoplasmic protein toxicity. Thus, closer examination of protein toxicity in the organelles in which toxic proteins tend to accumulate is warranted.
Protein toxicity in the mitochondria
The importance of the mitochondria to cell survival can easily be envisaged, as they are the organelles primarily responsible for ATP production in eukaryotic cells. Thus, mitochondrial dysfunction can be detrimental for cell survival, which can be catastrophic particularly to the brain, for the following reasons. First, most neurons cannot be replaced and thus need to be maintained due to their post-mitotic nature. This will inevitably lead to the accumulation of mitochondrial toxicity, by which the irreplaceable neurons will eventually succumb to death. Second, the excitability of neurons allows for significant influx of calcium ions that are buffered by mitochondria, the dysfunction of which will lead to excitotoxicity. Third, the elongated morphology of neurites entails a local supply of ATP by the mitochondria, the dysfunction of which will perturb growth and maintenance of neurites [110]. Hence, it is not surprising that mitochondrial dysfunction is one of the cardinal features of neurodegenerative diseases.
Mitochondrial dysfunction can be both primary and secondary drivers of neurodegeneration. In this section, we will mainly deal with the cases in which mitochondrial dysfunction is clearly a direct primary consequence of protein toxicity in the mitochondria. The following six toxic disease proteins that accumulate in mitochondria will be discussed: amyloid beta, amyloid precursor protein (APP), α-synuclein, mutant htt, TDP-43, and poly-GR DPRs.
Extracellular amyloid beta accumulation is one of the key pathological hallmarks of AD, in which mitochondrial dysfunction is often observed [111]. No direct mechanistic link between amyloid beta and mitochondrial dysfunction was identified until Lustbader et al. showed in 2004 that amyloid beta can localize to the mitochondria and directly bind to amyloid beta-binding alcohol dehydrogenase (ABAD) to induce mitochondrial toxicity [112]. Amyloid beta has also been shown to interact with cyclophilin D (CypD), an integral component of the mitochondrial permeability transition pore (mPTP), which sensitizes the opening of mPTP in both AD patients and mAPP mice brains [113].
APP, from which amyloid beta is derived, has also been shown to produce mitochondrial toxicity in models of, and patients with, AD. Anandatheerthavarada and colleagues showed that APP has a leader sequence with which APP localizes to the mitochondria in HCN-1A cells. The large acidic domain residues of APP (220–290) were found to clog the pores of TOM40 and TIM23, mitochondrial translocase of outer and inner membrane, respectively [114, 115]. When the authors experimented with postmortem human brain samples, they found that mitochondrial APP was observed only in AD brains [115]. Why APP does not localize to mitochondria under normal condition is currently unknown. In any case, this evidence strongly suggests that physical interaction of APP and amyloid beta with mitochondrial proteins is sufficient to generate oxidative stress, reduce ATP production, depolarize mitochondrial membrane potential, and sensitize mPTP opening, all of which contribute strongly to the mitochondrial dysfunction manifested in AD [110, 111, 116]. A recent study that shows reduction in amyloid beta toxicity by promoting mitochondrial proteostasis underscores the contribution of mitochondrial dysfunction in AD pathogenesis [117].
Mitochondrial dysfunction is not unique to AD. In HD, an energy-deficit related to mitochondrial dysfunction was first observed more than two decades ago [118]. The mechanisms by which mutant htt proteins induce mitochondrial dysfunction have been shown to be as diverse as that in AD. Aside from the mutant htt perturbing transcription of genes related to mitochondrial biogenesis and function in the nucleus [119, 120], it could also directly interact with mitochondrial proteins [121]. The N-terminal fragment of mutant htt localizes to the mitochondria [122–124] both in vivo and in vitro, and it interacts with the TIM23 complex, thereby clogging the mitochondrial import process [125]. These toxic interactions of mutant htt with mitochondrial proteins perturb calcium regulation, sensitize mPTP opening, depolarize mitochondrial membrane potential, and ultimately lead to neuronal demise [122–125].
Many genetic mutations linked to PD have been shown to cause mitochondrial dysfunction [126]. α-Synuclein, which is the central aggregating component of the Lewy bodies found in PD and Lewy body diseases, has high affinity for negatively charged lipids, including mitochondrial membranes [127–129]. In addition, α-synuclein has been shown to bind to several mitochondrial proteins such as the voltage-dependent anion channel (VDAC) in a monomeric form [129] and to TOM20 in an oligomeric form [130]. These interactions hinder the exchange of ATP/ADP between the mitochondria and the cytosol and impair mitochondrial protein import, both of which undermine mitochondrial function [129, 130].
ALS and FTD are diseases that manifest different clinical symptoms and yet share overlapping etiology. The pathological hallmark of ALS/FTD is the cytoplasmic mis-localization of TDP-43, but the mechanism by which TDP-43 proteins cause toxicity in the cytoplasm remains unclear. Wang et al. proposed a novel mode of toxicity by showing that TDP-43 possesses internal mitochondrial targeting signals that can direct TDP-43 into the mitochondria. The mitochondrial targeting becomes enhanced in ALS or FTD patients, which perturbs oxidative phosphorylation by means of binding to mitochondria-transcribed ND3 and ND6 mRNA and prohibiting their translation [131]. Conversely, Kawamata et al. reported that disease-associated mutant TDP-43 (TDP43 A315T) expression did not lead to any aberrant mitochondrial functions aside from calcium dysregulation [132]. These conflicting data warrant further investigation for us to assess more accurately the potential relevance of the mechanism described above. Interestingly, one of the arginine-rich DPRs (poly-GR repeats) derived from the hexanucleotide expansion mutation of C9ORF72 has also been shown to localize to the mitochondria and interact with mitochondrial ribosomal proteins, thereby causing mitochondrial dysfunction [133]. These recent findings suggest the mitochondria to be the primary driver of neurodegeneration in ALS/FTD as well.
How and for what purpose do these disease-associated toxic proteins accumulate in the mitochondria? Such heterogeneity of disease-associated proteins targeting mitochondria suggests non-specific mechanisms in which mitochondria act as cellular waste bins for toxic and presumably misfolded disease proteins. Ruan et al. recently proposed, in a rather timely manner, the mechanism by which misfolded cytoplasmic proteins accumulate inside the mitochondria to be degraded [134]. Ruan et al. showed that upon heat stress, misfolded cytoplasmic proteins enter mitochondria via mitochondrial translocase Tom70/Tom40 and are degraded by Pim1 in yeast [134]. Given that most toxic disease proteins are prone to misfolding, the potential relevance of this mechanism may be far-reaching in understanding the mitochondrial pathology common to most neurodegenerative diseases.
Protein toxicity in the stress granules
Neurons undergoing degeneration display immense stress to which multifaceted responses are launched to mitigate it. One of the key processes that occur in response to cellular stress is the formation of stress granules (SGs) [135]. Upon stress induction, cap-dependent translational processes are aborted and the messenger ribonucleoproteins (mRNPs) disengaged from the ribosomes begin to coalesce [136]. The RNA-binding proteins (RBPs) in these mRNPs interact electrostatically with one another through low complexity domains (LCDs) [137]. These interactions eventually facilitate liquid–liquid phase separation (LLPS) from the cytoplasm, thereby forming SGs. Concomitant to the formation of SGs, chaperones such as HSP70 are up-regulated via m6A-mediated cap-independent translation [138] to defuse stress by promoting refolding or degradation of misfolded proteins. Once the stress is resolved, the chaperones [139], along with autophagy [140], become instrumental in the disassembly of SGs [141].
In some neurodegenerative diseases, such as ALS or FTD, the SGs are infiltrated by disease-associated proteins that elicit an improper stress response. Many of the ALS genes encode proteins that are associated with SGs, such as Profilin-1, hnRNPA1/A2, fused in sarcoma (FUS), T cell-restricted intracellular antigen-1 (TIA1), and TDP-43, several of which are also linked to FTD [135, 142–147]. Increased cytoplasmic concentration or a mutation in the LCD of these proteins seems conducive to the initial LLPS, with subsequent stabilizing effect of SGs beyond the physiological necessity. The stabilized SGs that persist may then evolve into pathological fibrils [141].
Other ALS or FTD-associated proteins, such as superoxide dismutase-1 (SOD1) and valosin-containing protein (VCP), also impinge on the SGs, albeit by mechanisms that are independent of LCD [136, 148]. SOD1 variants associated with ALS form aggregates around the SGs, which suggests that SG formation precedes SOD1 aggregation [148]. Encapsulated by mutant SOD1, SGs display reduced dynamics and irregular morphology [148]. Such perturbed SG dynamics can be effectively combatted by the PQC system, of which VCP is a prominent member [136]. VCP is an ubiquitin segregase that uses ATP to extract ubiquitinated proteins from complexes to which they belong [136]. Stress induces SUMOylation of the VCP N-terminal domain by Ubc9, and it is one of the mechanisms by which VCP localizes to the SGs [149]. Mutations of VCP in the N-terminal domain thus have been shown to hinder SUMOylation, the modification without which hampers VCP from infiltrating SGs to extract ubiquitinated misfolded proteins for degradation [149]. Hence, SG disassembly fails with VCP mutation and the property of these SGs slowly transforms from dynamic liquid droplet like to pathological fibrils.
Whether or not SG pathology is associated with HD is still controversial. One study showed that mutant htt forms stress bodies, but not SGs [150]. Another study showed that both normal and mutant htt proteins can bind to SG-related factors such as Caprin-1 and G3BP1 [151]. Under normal conditions, neither normal nor mutant htt co-localized with the SG marker TIA1, whereas after arsenite treatment, both normal and mutant htt co-localized with TIA1; no difference in SG dynamics could be observed between normal and mutant htt in striatal precursor cells with or without arsenite treatment [151]. Similarly, another study reported that both normal and mutant htt interact with Caprin-1 and G3BP1; however, this study showed that the size and number of SGs were larger in striatal precursor cells expressing mutant htt compared to cells expressing normal htt [152]. These studies focused on mis-localization of mutant htt into SG to disrupt its dynamics; the results of these studies were mixed. Some studies suggest that rather than mutant htt localizing to SGs to cause their dysfunction, SG-related factors may mis-localize to mutant htt inclusions [153]. Time-lapse images in AD293 cells showed the formation of mutant htt inclusions with subsequent TIA-1 recruitment [154]. Another study showed that less than 1% of the interactors of mutant htt also interacted with SGs [155], which seems to support the view that mutant htt inclusions recruit certain SG-related factors.
Although the link between SG pathology and neurodegeneration has been well established, there is a lack of literature on the mechanism by which pathological SGs precipitate neurodegeneration. It has been a commonly accepted notion that SG formation upon stress induction contributes substantially to the global translational shutdown. However, a recent study using RNA-sequencing and single-molecule fluorescence in situ hybridization (smFISH) showed that only 10–12% of total mRNA molecules are localized to SGs [156], which does not support the notion that SGs are indispensable for global suppression of translation. Indeed, a previous study showed global translational shutdown upon stress induction in cells with G3BP mutations that prohibit SG formation [157]. This evidence supports the notion that SGs are dispensable for global suppression of translation. If it is not global translational shutdown, what then is the major function of SGs during stress, and how do pathological SGs precipitate neurodegeneration? Several studies showed that SG formation could impinge on intracellular signaling by sequestering key signaling molecules such as mammalian target of rapamycin (mTORC1) [135, 158, 159]. Thus, one of the mechanisms by which pathological SGs precipitate neurodegeneration may be through chronic impediment of intracellular signaling. Henceforth, elucidating the mechanistic link between pathological SGs and neurodegeneration should be one of the major focal points of research.
Propagation of toxic disease proteins
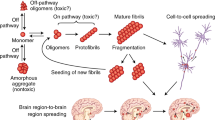
One of the interesting features often observed in neurodegenerative diseases is the gradual expansion of brain regions affected by pathogenic protein aggregates over time. In postmortem brains of PD patients, histopathological analyses have revealed the stereotypical progression of pathogenic inclusions from the autonomic nervous system, and from the dorsal motor and anterior olfactory nuclei to the substantia nigra, basal forebrain and the locus coeruleus, as well as to the hippocampus, neocortex, and basal ganglia [160] (Fig. 2a). In the postmortem brains of AD patients, tau inclusions initially appear in the transentorhinal cortex and later emerge in the hippocampal formation and neocortex [161] (Fig. 2b). These observations have led to an intriguing hypothesis that the expansion of the damaged brain regions is due to the gradual “prion-like” intercellular transmission of aggregates rather than the cell-autonomous accumulation of neuronal aggregates [162–164]. Supporting this hypothesis, clinical studies have shown that healthy embryonic mesencephalic neurons implanted into the striatum of patients with advanced PD developed scattered α-synuclein- and ubiquitin-positive inclusions many years after transplantation [165]. Similarly, healthy neurons implanted into the striatum of transgenic mice overexpressing human α-synuclein exhibited an accumulation of Lewy body-like inclusions [166–169]. In addition, the implementation of either patient-derived fibroblasts or pluripotent stem cells carrying mutant htt into the brain of neonatal wild-type mice was shown to induce cell-to-cell propagation of the mutant protein, a progressive loss of host cells, and behavioral deficits characteristic to HD [170]. These results suggest that pathological aggregates can transfer between diseased and healthy cells in humans and animals.

Propagation of misfolded proteins via intercellular transmission. Gradual change in the distribution of α-synuclein (a) and tau (b) inclusions in the brains of patients suffering from Parkinson’s (a) and Alzheimer’s (b) diseases. c Intercellular transmission of misfolded proteins via exocytosis, endocytosis, exosomes, and tunneling nanotubes
The more direct evidence supporting the mobility of aggregates between cells has been provided by studies either employing an intracerebral application of exogenous aggregates derived from diseased humans and animals or ectopic overexpression of α-synuclein and tau in a population of neurons to examine whether the aggregates can spread through the brain connectome [166, 171–173]. For example, an intracerebral injection of brain extracts prepared from the symptomatic P301S tau transgenic mice was shown to be sufficient for inducing neurofibrillary tangles in presymptomatic P301S tau transgenic mice at the injection site as well as in the distant brain regions that are physically separated by one or more synapses from the injection site [166, 174]. Similarly, within 6–12 months after the inoculation of amyloid beta-containing brain extracts derived from either AD patients or aged APP transgenic mice into the hippocampus and neocortex of young APP transgenic mice, amyloid beta deposition and its associated pathology were widespread in the brain [175–177]. Likewise, an intracerebral administration of brain or spinal cord homogenates prepared from symptomatic α-synuclein transgenic mice facilitated the appearance and spread of Lewy pathology in presymptomatic recipient transgenic mice [171]. The spread of pathological changes was recapitulated by a local injection of synthetic α-synuclein fibrils or tau filaments in presymptomatic transgenic mice, suggesting that aggregates, but not other factors in the brain homogenates, are sufficient for the spreading of the pathological changes in the brain [171, 178–180]. Finally, selective overexpression of transgenic tau, amyloid beta, or α-synuclein in a population of neurons could trigger the spread of misfolded proteins to the interconnected brain regions in transgenic mice [181–186].
Several lines of evidence suggest that peripherally introduced aggregates can lead to the accumulation of misfolded proteins in the central nervous system (CNS). For instance, transgenic mice expressing a mutant human α-synuclein exhibited pathogenic inclusions and neuroinflammatory responses throughout the CNS within 2–4 months after an intramuscular injection of recombinant α-synuclein fibrils. Those animals also displayed a debilitating motor impairment, which is one of the clinical symptoms characteristic of PD [187]. However, when the sciatic nerve, which connects the muscles to the spinal cord, is severed in those same mice, the development of the pathogenic inclusions and neuroinflammatory responses in the CNS was significantly delayed. This suggests that the retrograde transport of misfolded proteins via the peripheral nerve is required for disease propagation, at least in this mouse model. A recent study has shown that α-synuclein fibrils injected into the olfactory bulb of wild-type mice propagate transneuronally to distant brain regions and induce progressive olfactory deficits [188]. Similarly, mutant htt ectopically expressed in sensory receptor neurons in Drosophila can spread transcellularly to neuronal and glial cells in the brain [189–191]. Another study has demonstrated that an intestinal application of either the brain lysate from human PD patients or recombinant α-synuclein in rats could elicit α-synuclein inclusions in the dorsal motor nucleus of the vagus nerve in the brainstem [192]. Moreover, systemic treatment of aggregates, such as repeated injections of α-synuclein fibrils into the tail vein and an intraperitoneal inoculation of tau extracts or amyloid beta seeds, was sufficient to cause accumulation of deposits in the brain [193–196].
There are several varying molecular mechanisms by which pathogenic aggregates can transfer between cells (Fig. 2c). Exocytosis is one of the main secretory mechanisms involved in releasing aggregates from donor cells, which occurs in an intracellular calcium- and endosome-dependent manner [197–199]. Alternatively, the misfolded proteins can be released into the extracellular space within secretory vesicles called exosomes [200–202]. Exosomes are vesicles of 50–100 nm diameter that normally mediate intercellular transportation of mRNA, small regulatory RNA, and specific proteins between the cells [203]. A number of studies have demonstrated that exosome-mediated propagation is implicated in the spreading of pathogenic inclusions in neurodegenerative diseases. Studies using immunofluorescence and immunoelectron microscopy have revealed that the exosomes are associated with amyloid beta peptides, phosphorylated tau, and other related molecules [204]. Furthermore, exosomes isolated from diseased transgenic animals or human patients were shown to have an ability to nucleate oligomerization of endogenous proteins in recipient cells [205, 206]. Accordingly, pharmacological inhibition of key regulatory enzymes mediating secretion and synthesis of the exosomes significantly reduced both the amyloid plaque formation in the AD mouse model and the secretion and propagation of tau from microglia in vitro and in vivo [182, 207]. Finally, pathogenic inclusions can be transferred through tunnel-like structures called tunneling nanotubes that connect the cytosolic compartments between neighboring cells to facilitate intercellular material exchange for communication [208]. The diameter of nanotubes ranges from 50 to 200 nm, and their lengths can reach up to several cell diameters [209]. In vitro studies have shown that α-synuclein fibrils can be transferred via tunneling nanotubes to lysosomes of recipient cells, such as mouse catecholaminergic cells and human primary brain pericytes, and subsequently induce the aggregation of cytosolic α-synuclein [208, 210].
In sum, the progressive accumulation of specific protein aggregates along anatomical connections is a common hallmark of major neurodegenerative diseases such as AD and PD. Extensive evidence from in vitro and in vivo studies suggests that one of the fundamental pathogenic mechanisms by which neurodegeneration transpires is the intercellular transmission of protein aggregates in synaptically connected brain networks.
As describe above, disease propagation model is supported by a number of preclinical evidence, but there are also some observations that cannot be fully explained by this model. For example, a fetal graft implanted in some PD patients was found to be without pathology in autopsies performed two decades following transplantation [211, 212]. In addition, proteins associated with neurodegenerative diseases are unlikely to transmit between individuals as a disease-causing infectious agent [213, 214]. Finally, a recent study showed that brain regions manifesting Lewy pathology neither fully correlate with the synaptic connection patterns revealed by connectome mapping [215] nor follow the spatiotemporal spread patterns described by Braak et al. Thus, further research is required to fully understand the clinical relevance of the aggregate propagation model versus the cell-autonomous model.
Selective neuronal vulnerability
Most of the genes whose mutations cause neurodegenerative diseases are ubiquitously expressed in all developmental stages of life. However, developmental defects are minimal in patients with neurodegenerative diseases such as AD, HD, PD, or ALS. In addition, neurodegenerative disease patients tend to manifest late-onset, cell-type-specific neurodegeneration [216]. Due to their post-mitotic nature, neurons may be more vulnerable to cellular toxicity than other cell types which are capable of regeneration. Furthermore, neurons are generally more ATP dependent than other cell types, rendering neurons more vulnerable to energy crises caused by membrane potential changes and mitochondrial dysfunction. Nevertheless, two important questions remain to be answered. First, what accounts for the selective neuronal toxicity? Second, why does such toxicity stay dormant during development, but become damaging with age?
Both sporadic and familial disease cases present with selective neuronal vulnerability. This selective neuronal vulnerability is often indistinguishable between patients with sporadic and familial etiology, but is distinct from disease to disease [217]. Hence, we speculate that selective neuronal vulnerability may arise from genetic predisposition or environmental factors that chiefly affect certain neurons. For instance, PD is often associated with mutations in genes that are involved in mitochondrial function and also with exposure to environmental mitochondrial toxins [126]. Whether caused by genetic or environmental factors, PD involves selective degeneration of the substantia nigra pars compacta (SNpc). Thus, we deduce that the SNpc may be particularly vulnerable to mitochondrial dysfunction. What makes SNpc especially vulnerable to mitochondrial dysfunction is unclear, though the unique properties of those neurons, such as the oxidation of dopamine neurotransmitters and the pacemaking activity of Cav1.3 L-type Ca2+ channels [218], are likely contributors. Nonetheless, the possibility of other factors contributing to the selective SNpc degeneration should not be excluded.
Similarly, both sporadic and familial ALS are associated with RNA metabolism [219], the dysfunction of which may selectively render upper and lower motor neurons vulnerable to degeneration. Interestingly, RNA metabolism is also compromised by the activation of human endogenous retrovirus k [220], which is associated with ALS [221]. Hence, RNA metabolism dysfunction may be associated with ALS, but whether it can cause selective motor neuron degeneration is still unclear.
PolyQ disease patients tend to exhibit cerebellar atrophy [222]. This outcome suggests that the cerebellum is particularly vulnerable to protein toxicity mediated by the expanded polyQ proteins. We speculate that the cerebellum may have a weaker defense system against polyQ toxicity or that it expresses a disproportionate amount of proteins that are polyQ targets. Interestingly, fetal alcohol exposure primarily causes cerebellar pathology, which is linked to reduced CBP expression in the cerebellum [223]. In addition, Rubinstein–Taybi syndrome, which is caused by a CBP loss-of-function mutation, involves cerebellar pathology [224]. Since many different polyQ proteins have been shown to sequester and to interfere with CBP [225, 226], we speculate that a polyQ-induced loss of CBP function may contribute to the selective cerebellar pathology in polyQ diseases.
There are many possible explanations for minimal developmental defects in patients who later develop neurodegenerative diseases. We believe that the following are the three most viable explanations: (1) the PQC system may mitigate protein toxicity early in life but may fail later in life, (2) protein toxicity eventually reaches a critical threshold, beyond which defense mechanisms start to collapse, or (3) environmental factors or epigenetic alterations during and after development contribute to the disease onset later in life. We believe that all of these processes may contribute to the late onset of neurodegenerative diseases. Thus, we propose that development is the critical window within which therapeutics should be applied to prevent or delay disease initiation.
Discussion and future perspectives
Neurodegenerative diseases, for which there are no remedies, correlate well with age, and this is a major conundrum with which we are confronted in an aging society. With years of massive research efforts carried out in laboratories around the globe, much knowledge of the nature of neurodegenerative diseases has been accrued with only a minimal progress in the actual development of effective therapeutics. To bridge the gap between our current understanding of the disease and the application thereof to the development of effective therapeutics, in this review, we have systematically analyzed and summarized the mechanistic underpinnings of protein toxicity (Table 1; Fig. 1), which is central to the development and progression of a vast array of neurodegenerative diseases such as AD, PD, ALS, FTD, and HD. We have discussed a number of toxic disease proteins within their respective subcellular contexts in an attempt to compare and contrast their pathogenic mechanisms in a localized area.
In this review, we have focused on the mechanisms of protein toxicity in neurodegenerative diseases, but protein toxicity can also be observed in psychiatric disorders such as schizophrenia. Recently, schizophrenia has been associated with genes such as Neuronal PAS Domain Protein 3 (NPAS3), Disrupted-in-schizophrenia 1(DISC1), and TRIO binding protein-1 (TRIOBP-1); translocation or point mutations in these genes may cause protein aggregation [227–229]. NPAS3-V304I proteins form aggregates, into which normal NPAS3 proteins are sequestered; NPAS3 loss of function then leads to decreased transcription of its downstream target, VGF [228]. A DISC1 translocation mutation produces a truncated DISC1 protein, which can form aggregates and can act in a dominant negative manner. Three polymorphisms of DISC1 have also been associated with major depression and schizophrenia [229]. TRIOBP-1 has been found in insoluble aggregates within brain lysates of schizophrenia patients’ brains. Amino acids 324–348 of TRIOBP-1 are thought to be critical for aggregation; TRIOBP-1 aggregation may affect actin dynamics and neurite growth [230]. Interestingly, the TRIOBP mutation is associated with deafness, which is often associated with psychiatric disorders. One study identified a family with schizophrenia and hearing impairment; for this family, the locus in which the causative mutation lies includes TRIOBP [231]. In addition to NPAS3, DISC1, and TRIOBP-1, CRMP1 and dysbindin can also form protein aggregates in schizophrenic patients [229, 232]; therefore, we infer that protein toxicity may be one of the mechanisms by which schizophrenia occurs. Interestingly, schizophrenia has been suggested to be linked to polyQ diseases as well [233]. It has been shown biochemically that DISC1 binds to mutant htt more strongly than it binds to normal htt [234]. This binding sequestered DISC1 away from PDE4, thereby increasing its activity. Overexpressing modified DISC1, which can interact with PDE4 but not with mutant htt, ameliorated anhedonia in a mouse model of HD [234]; anhedonia is one of the core features of schizophrenia. Many neurodegenerative disease patients also display mental or psychiatric symptoms such as depression and hallucinations [235, 236]; however, the molecular link between neurodegeneration and psychiatric symptoms remain undefined.
Increased life expectancy and the prevalence of neurodegenerative diseases in the twenty-first century are driving therapeutic research. However, currently there are only palliative drugs available to treat these diseases. The task of drug development is formidable; it has been estimated that AD drug development efforts face a 99.6% failure rate [237]. Hence, Pfizer (one of the leading pharmaceutical companies) recently announced its exit from the field of neuroscience [238]. However, as basic research is slowly helping us to understand the complexity of the brain, new treatment strategies against neurodegenerative diseases are being formulated.
One of the fastest-growing treatment strategies is antibody utilization [239]. For neurodegenerative diseases that involve protein toxicity, elimination of toxic proteins is an efficient way in which toxicity can be mitigated. Hence, antibodies against toxic disease proteins such as α-synuclein and amyloid beta are being developed. Recently, Biogen Inc. developed aducanumab, which was shown to reduce both amyloid plaques and cognitive decline in patients with mild form of AD after a 12-month trial [240]. However, another amyloid beta antibody (solanezumab) did not mitigate cognitive decline or reduce amyloid plaque in AD patients [241]. There are a few explanations that may account for this discrepancy. First, solanezumab administration may have been below the effective dose. Second, solanezumab may have bound to the wrong target. Third, the disease of patients in the solanezumab study may have been too advanced for the treatment to have been beneficial. Although only approximately 0.1% of the antibodies are known to traverse the blood–brain barrier [239], intravenous infusion of 400 mg every 4 weeks [241] seems to be a substantial dosage. Solanezumab targets amyloid beta monomers [241], whereas aducanumab targets oligomers and fibrils [240]; recent studies suggest that the oligomeric form may be the most toxic form [242]. This suggests that therapeutic target may have been at fault. Nevertheless, disease progression could perhaps be delayed if solanezumab was administered before any substantial oligomers or fibrils were formed. In any case, we can learn from these two examples, which highlight the significance of identifying the key drug target, correct dosage, and the disease stage at which to intervene.
Antibody treatment has its own drawbacks, however. As they cannot freely traverse across membranes, intracellular targeting of antibodies is very difficult, and intra-organellar targeting, even more so. Thus, antibody-based treatments have been more successful with extracellular targets (such as amyloid plaques) instead of intracellular targets (such as mutant htt and α-synuclein). Nevertheless, there are a few notable antibody-based drugs (RO7046015 from Roche and BIIB054 from Biogen) undergoing clinical tests targeting cell-to-cell transmission of α-synuclein [243]. Although there are various methods whereby antibody-based drugs can be delivered intracellularly in vitro and ex vivo [239], delivery in vivo often still poses insurmountable challenges. Hence, we believe that undertaking the challenge of target-specific delivery will be crucial in advancing the development of effective therapeutics against neurodegenerative diseases.
Our review discussed the mechanisms and the sites at which protein toxicity occurs to assist in the identification of druggable targets. We have also briefly discussed potential mechanisms of cell-to-cell propagation of toxic proteins and selective neuronal vulnerability in neurodegenerative diseases. We hope that by enhancing our understanding in these areas of research, more effective therapeutic strategies will be developed in the future.
References
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296(5575):1991–1995. https://doi.org/10.1126/science.1067122
Flagmeier P, Meisl G, Vendruscolo M, Knowles TP, Dobson CM, Buell AK, Galvagnion C (2016) Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of alpha-synuclein aggregation. Proc Natl Acad Sci USA 113(37):10328–10333. https://doi.org/10.1073/pnas.1604645113
Hunter S, Brayne C (2018) Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatry 23(1):81–93. https://doi.org/10.1038/mp.2017.218
Neumann M (2009) Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci 10(1):232–246. https://doi.org/10.3390/ijms10010232
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338. https://doi.org/10.1126/science.1232927
Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2017.00035
Shao J, Diamond MI (2007) Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet 16(Spec No 2):R115–R123. https://doi.org/10.1093/hmg/ddm213
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9(10):995–1007. https://doi.org/10.1016/S1474-4422(10)70195-2
Klein C, Westenberger A (2012) Genetics of parkinson's disease. Cold Spring Harb Perspect Med 2(1):a008888–a008888
Bekris LM, Yu CE, Bird TD, Tsuang DW (2010) Review Article: Genetics of Alzheimer Disease. J Geriatr Psychiatry Neurol 23(4):213–227
Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26(34):8774–8786. https://doi.org/10.1523/JNEUROSCI.2315-06.2006
Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA (2005) Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA 102(1):238–243. https://doi.org/10.1073/pnas.0406173102
Woulfe J (2008) Nuclear bodies in neurodegenerative disease. Biochem Biophys Acta 1783(11):2195–2206. https://doi.org/10.1016/j.bbamcr.2008.05.005
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431(7010):805–810. https://doi.org/10.1038/nature02998
Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416(6880):507–511. https://doi.org/10.1038/416507a
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618):486–489. https://doi.org/10.1126/science.1079469
Woulfe JM (2007) Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol 33(1):2–42. https://doi.org/10.1111/j.1365-2990.2006.00819.x
Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY (2005) Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin–proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14(5):679–691. https://doi.org/10.1093/hmg/ddi064
Kim HJ, Taylor JP (2017) Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96(2):285–297. https://doi.org/10.1016/j.neuron.2017.07.029
Kwon MJ, Kim S, Han MH, Lee SB (2016) Epigenetic changes in neurodegenerative diseases. Mol Cells 39(11):783–789. https://doi.org/10.14348/molcells.2016.0233
Havel LS, Li S, Li XJ (2009) Nuclear accumulation of polyglutamine disease proteins and neuropathology. Mol Brain 2:21. https://doi.org/10.1186/1756-6606-2-21
Pouladi MA, Morton AJ, Hayden MR (2013) Choosing an animal model for the study of Huntington’s disease. Nat Rev Neurosci 14(10):708–721. https://doi.org/10.1038/nrn3570
Huynh DP, Figueroa K, Hoang N, Pulst SM (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet 26(1):44–50. https://doi.org/10.1038/79162
Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291(5512):2423–2428. https://doi.org/10.1126/science.1056784
Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97(12):6763–6768. https://doi.org/10.1073/pnas.100110097
McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH (2000) CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9(14):2197–2202
Strom AL, Forsgren L, Holmberg M (2005) A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis 20(3):646–655. https://doi.org/10.1016/j.nbd.2005.04.018
Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, Gusella JF (1998) Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet 24(4):217–233
Boutell JM, Thomas P, Neal JW, Weston VJ, Duce J, Harper PS, Jones AL (1999) Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum Mol Genet 8(9):1647–1655
Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35(1):76–83. https://doi.org/10.1038/ng1219
Kwon MJ, Kim JH, Kim T, Lee SB (2017) Pharmacological intervention of early neuropathy in neurodegenerative diseases. Pharmacol Res 119:169–177. https://doi.org/10.1016/j.phrs.2017.02.003
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413(6857):739–743. https://doi.org/10.1038/35099568
Li FS, Macfarlan T, Pittman RN, Chakravarti D (2002) Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem 277(47):45004–45012. https://doi.org/10.1074/jbc.M205259200
Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, Fraenkel E (2013) Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci USA 110(6):2354–2359. https://doi.org/10.1073/pnas.1221292110
Thomas B, Matson S, Chopra V, Sun L, Sharma S, Hersch S, Rosas HD, Scherzer C, Ferrante R, Matson W (2013) A novel method for detecting 7-methyl guanine reveals aberrant methylation levels in Huntington disease. Anal Biochem 436(2):112–120. https://doi.org/10.1016/j.ab.2013.01.035
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254. https://doi.org/10.1038/ng1089
Chung CG, Kwon MJ, Jeon KH, Hyeon DY, Han MH, Park JH, Cha IJ, Cho JH, Kim K, Rho S, Kim GR, Jeong H, Lee JW, Kim T, Kim K, Kim KP, Ehlers MD, Hwang D, Lee SB (2017) Golgi outpost synthesis impaired by toxic polyglutamine proteins contributes to dendritic pathology in neurons. Cell Rep 20(2):356–369. https://doi.org/10.1016/j.celrep.2017.06.059
Lee SB, Bagley JA, Lee HY, Jan LY, Jan YN (2011) Pathogenic polyglutamine proteins cause dendrite defects associated with specific actin cytoskeletal alterations in Drosophila. Proc Natl Acad Sci USA 108(40):16795–16800. https://doi.org/10.1073/pnas.1113573108
Kweon JH, Kim S, Lee SB (2017) The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep 50(1):5–11
Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, Zoghbi HY (2011) Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 334(6056):690–693. https://doi.org/10.1126/science.1212673
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 23(28):9418–9427
Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF (2005) Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem 280(1):556–563. https://doi.org/10.1074/jbc.M410210200
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA 100(4):2041–2046. https://doi.org/10.1073/pnas.0437870100
Ying MY, Xu R, Wu XH, Zhu HX, Zhuang Y, Han M, Xu T (2006) Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem 281(18):12580–12586. https://doi.org/10.1074/jbc.M511677200
Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA 101(7):2173–2178. https://doi.org/10.1073/pnas.0308512100
Ho L, Guo Y, Spielman L, Petrescu O, Haroutunian V, Purohit D, Czernik A, Yemul S, Aisen PS, Mohs R, Pasinetti GM (2001) Altered expression of a-type but not b-type synapsin isoform in the brain of patients at high risk for Alzheimer’s disease assessed by DNA microarray technique. Neurosci Lett 298(3):191–194
Wu ZL, Ciallella JR, Flood DG, O’Kane TM, Bozyczko-Coyne D, Savage MJ (2006) Comparative analysis of cortical gene expression in mouse models of Alzheimer’s disease. Neurobiol Aging 27(3):377–386. https://doi.org/10.1016/j.neurobiolaging.2005.02.010
Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB (2004) Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm (Vienna) 111(12):1543–1573. https://doi.org/10.1007/s00702-004-0212-1
Noureddine MA, Li YJ, van der Walt JM, Walters R, Jewett RM, Xu H, Wang T, Walter JW, Scott BL, Hulette C, Schmechel D, Stenger JE, Dietrich F, Vance JM, Hauser MA (2005) Genomic convergence to identify candidate genes for Parkinson disease: SAGE analysis of the substantia nigra. Mov Disord 20(10):1299–1309. https://doi.org/10.1002/mds.20573
Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L (2016) Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol 132(2):159–173. https://doi.org/10.1007/s00401-016-1586-5
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525(7567):56–61. https://doi.org/10.1038/nature14973
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525(7567):129–133. https://doi.org/10.1038/nature14974
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS (2016) Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351(6269):173–176. https://doi.org/10.1126/science.aad2033
Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C (2017) Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94(1):48–57 e44. https://doi.org/10.1016/j.neuron.2017.03.027
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, Pham JT, Ahmed I, Peng Q, Wadhwa H, Pletnikova O, Troncoso JC, Duan W, Snyder SH, Ranum LP, Thompson LM, Lloyd TE, Ross CA, Rothstein JD (2017) Mutant huntingtin disrupts the nuclear pore complex. Neuron 94(1):93–107 e106. https://doi.org/10.1016/j.neuron.2017.03.023
Boeynaems S, Bogaertl E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van den Bosch L (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep UK. https://doi.org/10.1038/srep20877
Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, McKnight SL (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci USA 114(7):E1111–E1117. https://doi.org/10.1073/pnas.1620293114
Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147. https://doi.org/10.1038/emm.2014.117
Deriziotis P, Andre R, Smith DM, Goold R, Kinghorn KJ, Kristiansen M, Nathan JA, Rosenzweig R, Krutauz D, Glickman MH, Collinge J, Goldberg AL, Tabrizi SJ (2011) Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J 30(15):3065–3077. https://doi.org/10.1038/emboj.2011.224
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin–proteasome system. Am J Pathol 181(4):1426–1435. https://doi.org/10.1016/j.ajpath.2012.06.033
Dantuma NP, Bott LC (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci 7:70. https://doi.org/10.3389/fnmol.2014.00070
Ju JS, Miller SE, Hanson PI, Weihl CC (2008) Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem 283(44):30289–30299. https://doi.org/10.1074/jbc.M805517200
Lee JS, Seo TW, Yi JH, Shin KS, Yoo SJ (2013) CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G. Cell Death Dis 4:e666. https://doi.org/10.1038/cddis.2013.181
Lu B, Al-Ramahi I, Valencia A, Wang Q, Berenshteyn F, Yang H, Gallego-Flores T, Ichcho S, Lacoste A, Hild M, Difiglia M, Botas J, Palacino J (2013) Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci 16(5):562–570. https://doi.org/10.1038/nn.3367
Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL (2005) CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci 25(40):9152–9161. https://doi.org/10.1523/JNEUROSCI.3001-05.2005
Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G (2007) CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci 27(19):5115–5126. https://doi.org/10.1523/JNEUROSCI.1242-07.2007
Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, Zhang YJ, Jiang H, Ross CA, Moore DJ, Patterson C, Petrucelli L, Dawson TM, Dawson VL (2009) CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci USA 106(8):2897–2902. https://doi.org/10.1073/pnas.0810123106
Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL (2009) In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis 33(3):342–353. https://doi.org/10.1016/j.nbd.2008.10.016
Bhat KP, Yan S, Wang CE, Li SH, Li XJ (2014) Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci USA 111(15):5706–5711. https://doi.org/10.1073/pnas.1402215111
Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L (2011) Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol 85(7):3120–3127. https://doi.org/10.1128/Jvi.02004-10
Lee JM, Wheeler VC, Chao MJ, Vonsattel JPG, Pinto RM, Lucente D, Abu-Elneel K, Ramos EM, Mysore JS, Gillis T, MacDonald ME, Gusella JF, Harold D, Stone TC, Escott-Price V, Han J, Vedernikov A, Holmans P, Jones L, Kwak S, Mahmoudi M, Orth M, Landwehrmeyer GB, Paulsen JS, Dorsey ER, Shoulson I, Myers RH, Dis GMH (2015) Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162(3):516–526. https://doi.org/10.1016/j.cell.2015.07.003
Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7:63. https://doi.org/10.3389/fnmol.2014.00063
Nucifora FC Jr, Nucifora LG, Ng CH, Arbez N, Guo Y, Roby E, Shani V, Engelender S, Wei D, Wang XF, Li T, Moore DJ, Pletnikova O, Troncoso JC, Sawa A, Dawson TM, Smith W, Lim KL, Ross CA (2016) Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat Commun 7:11792. https://doi.org/10.1038/ncomms11792
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. https://doi.org/10.1038/nn.3350
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295. https://doi.org/10.1126/science.1101738
Kabuta T, Furuta A, Aoki S, Furuta K, Wada K (2008) Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 283(35):23731–23738. https://doi.org/10.1074/jbc.M801918200
Jia H, Kast RJ, Steffan JS, Thomas EA (2012) Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum Mol Genet 21(24):5280–5293. https://doi.org/10.1093/hmg/dds379
Koga H, Cuervo AM (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 43(1):29–37. https://doi.org/10.1016/j.nbd.2010.07.006
Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH (2012) The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One 7(10):e46834. https://doi.org/10.1371/journal.pone.0046834
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13(5):567–576. https://doi.org/10.1038/nn.2528
Fu YH, Wu P, Pan YY, Sun XL, Yang HY, Difiglia M, Lu BX (2017) A toxic mutant huntingtin species is resistant to selective autophagy. Nat Chem Biol 13(11):1152. https://doi.org/10.1038/nchembio.2461
Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC (2010) Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol 21(7):691–698. https://doi.org/10.1016/j.semcdb.2010.02.008
Metcalf DJ, Garcia-Arencibia M, Hochfeld WE, Rubinsztein DC (2012) Autophagy and misfolded proteins in neurodegeneration. Exp Neurol 238(1):22–28. https://doi.org/10.1016/j.expneurol.2010.11.003
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997. https://doi.org/10.1038/nm.3232
Hochfeld WE, Lee S, Rubinsztein DC (2013) Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin 34(5):600–604. https://doi.org/10.1038/aps.2012.189
Pereira CMF (2013) Crosstalk between endoplasmic reticulum stress and protein misfolding in neurodegenerative diseases. ISRN Cell Biol 2013:22. https://doi.org/10.1155/2013/256404
Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A (2001) VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ 8(10):977–984. https://doi.org/10.1038/sj.cdd.4400907
Duennwald ML, Lindquist S (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev 22(23):3308–3319. https://doi.org/10.1101/gad.1673408
Leitman J, Hartl FU, Lederkremer GZ (2013) Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. https://doi.org/10.1038/ncomms3753
Fujita K, Nakamura Y, Oka T, Ito H, Tamura T, Tagawa K, Sasabe T, Katsuta A, Motoki K, Shiwaku H, Sone M, Yoshida C, Katsuno M, Eishi Y, Murata M, Taylor JP, Wanker EE, Kono K, Tashiro S, Sobue G, La Spada AR, Okazawa H (2013) A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat Commun. https://doi.org/10.1038/ncomms2828
Yang H, Li JJ, Liu S, Zhao J, Jiang YJ, Song AX, Hu HY (2014) Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Sci Rep UK. https://doi.org/10.1038/srep06410
Cairns NJ, Lee VM, Trojanowski JQ (2004) The cytoskeleton in neurodegenerative diseases. J Pathol 204(4):438–449. https://doi.org/10.1002/path.1650
McMurray CT (2000) Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ 7(10):861–865. https://doi.org/10.1038/sj.cdd.4400764
Bamburg JR, Bloom GS (2009) Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton 66(8):635–649. https://doi.org/10.1002/cm.20388
Bamburg JR, Bernstein BW, Davis RC, Flynn KC, Goldsbury C, Jensen JR, Maloney MT, Marsden IT, Minamide LS, Pak CW, Shaw AE, Whiteman I, Wiggan O (2010) ADF/Cofilin-actin rods in neurodegenerative diseases. Curr Alzheimer Res 7(3):241–250
Chevalier-Larsen E, Holzbaur EL (2006) Axonal transport and neurodegenerative disease. Biochem Biophys Acta 1762(11–12):1094–1108. https://doi.org/10.1016/j.bbadis.2006.04.002
Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, Johnson GV, Brady ST (2013) Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron 78(1):109–123. https://doi.org/10.1016/j.neuron.2013.01.036
Roy S, Zhang B, Lee VM, Trojanowski JQ (2005) Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol 109(1):5–13. https://doi.org/10.1007/s00401-004-0952-x
Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST (2009) Axonal transport defects in neurodegenerative diseases. J Neurosci 29(41):12776–12786. https://doi.org/10.1523/JNEUROSCI.3463-09.2009
Kanaan NM, Pigino GF, Brady ST, Lazarov O, Binder LI, Morfini GA (2013) Axonal degeneration in Alzheimer’s disease: when signaling abnormalities meet the axonal transport system. Exp Neurol 246:44–53. https://doi.org/10.1016/j.expneurol.2012.06.003
Brady ST, Morfini GA (2017) Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol Dis 105:273–282. https://doi.org/10.1016/j.nbd.2017.04.010
Williamson TL, Cleveland DW (1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2(1):50–56
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G (2010) Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA 107(47):20523–20528. https://doi.org/10.1073/pnas.1006869107
Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ (2016) Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet 25(12):2378–2392. https://doi.org/10.1093/hmg/ddw105
Song Y, Nagy M, Ni W, Tyagi NK, Fenton WA, Lopez-Giraldez F, Overton JD, Horwich AL, Brady ST (2013) Molecular chaperone Hsp110 rescues a vesicle transport defect produced by an ALS-associated mutant SOD1 protein in squid axoplasm. Proc Natl Acad Sci USA 110(14):5428–5433. https://doi.org/10.1073/pnas.1303279110
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH Jr (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13(11):1396–1403. https://doi.org/10.1038/nn.2660
Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40(1):25–40
Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST (2003) Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40(1):41–52
Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118(1):127–138. https://doi.org/10.1016/j.cell.2004.06.018
Abramov AY, Berezhnov AV, Fedotova EI, Zinchenko VP, Dolgacheva LP (2017) Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem Soc Trans. https://doi.org/10.1042/BST20170024
Spuch C, Ortolano S, Navarro C (2012) New insights in the amyloid-Beta interaction with mitochondria. J Aging Res 2012:324968. https://doi.org/10.1155/2012/324968
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304(5669):448–452. https://doi.org/10.1126/science.1091230
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14(10):1097–1105. https://doi.org/10.1038/nm.1868
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG (2003) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161(1):41–54. https://doi.org/10.1083/jcb.200207030
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26(35):9057–9068. https://doi.org/10.1523/JNEUROSCI.1469-06.2006
Swerdlow RH (2017) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. https://doi.org/10.3233/JAD-170585
Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J (2017) Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. https://doi.org/10.1038/nature25143
Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41(5):646–653. https://doi.org/10.1002/ana.410410514
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127(1):59–69. https://doi.org/10.1016/j.cell.2006.09.015
Farshbaf MJ, Ghaedi K (2017) Huntington’s disease and mitochondria. Neurotox Res 32(3):518–529. https://doi.org/10.1007/s12640-017-9766-1
Kim YE, Hosp F, Frottin F, Ge H, Mann M, Hayer-Hartl M, Hartl FU (2016) Soluble oligomers of PolyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol Cell 63(6):951–964. https://doi.org/10.1016/j.molcel.2016.07.022
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5(8):731–736. https://doi.org/10.1038/nn884
Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 28(11):2783–2792. https://doi.org/10.1523/JNEUROSCI.0106-08.2008
Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 13(14):1407–1420. https://doi.org/10.1093/hmg/ddh162
Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ, Kim AH, Friedlander RM (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17(6):822–831. https://doi.org/10.1038/nn.3721
Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ (2014) A mitocentric view of Parkinson’s disease. Annu Rev Neurosci 37:137–159. https://doi.org/10.1146/annurev-neuro-071013-014317
Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273(16):9443–9449. https://doi.org/10.1074/jbc.273.16.9443
Pfefferkorn CM, Jiang ZP, Lee JC (2012) Biophysics of alpha-synuclein membrane interactions. BBA-Biomembr 1818(2):162–171. https://doi.org/10.1016/j.bbamem.2011.07.032
Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC, Lee JC, Bezrukov SM (2015) Alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J Biol Chem 290(30):18467–18477. https://doi.org/10.1074/jbc.M115.641746
Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu XP, Mccoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT (2016) alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaf3634
Wang WZ, Wang LW, Lu JJ, Siedlak SL, Fujioka H, Liang JJ, Jiang SR, Ma XP, Jiang Z, da Rocha EL, Sheng M, Choi H, Lerou PH, Li H, Wang XL (2016) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22(8):869. https://doi.org/10.1038/nm.4130
Kawamata H, Peixoto P, Konrad C, Palomo G, Bredvik K, Gerges M, Valsecchi F, Petrucelli L, Ravits JM, Starkov A, Manfredi G (2017) Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol Neurodegener 12(1):37. https://doi.org/10.1186/s13024-017-0180-1
Lopez-Gonzalez R, Lu YB, Gendron TF, Karydas A, Tran H, Yang DJ, Petrucelli L, Miller BL, Almeida S, Gao FB (2016) Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92(2):383–391. https://doi.org/10.1016/j.neuron.2016.09.015
Ruan LH, Zhou CK, Jin EL, Kucharavy A, Zhang Y, Wen ZH, Florens L, Li R (2017) Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543(7645):443. https://doi.org/10.1038/nature21695
Mahboubi H, Stochaj U (2017) Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochem Biophys Acta 4:884–895. https://doi.org/10.1016/j.bbadis.2016.12.022
Protter DS, Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26(9):668–679. https://doi.org/10.1016/j.tcb.2016.05.004
Hyman AA, Weber CA, Julicher F (2014) Liquid–liquid phase separation in biology. Annu Rev Cell Dev Biol 30:39–58. https://doi.org/10.1146/annurev-cellbio-100913-013325
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR (2015) 5′ UTR m(6)A promotes cap-independent translation. Cell 163(4):999–1010. https://doi.org/10.1016/j.cell.2015.10.012
Walters RW, Muhlrad D, Garcia J, Parker R (2015) Differential effects of Ydj1 and Sis1 on Hsp70-mediated clearance of stress granules in Saccharomyces cerevisiae. RNA 21(9):1660–1671. https://doi.org/10.1261/rna.053116.115
Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153(7):1461–1474. https://doi.org/10.1016/j.cell.2013.05.037
Alberti S, Mateju D, Mediani L, Carra S (2017) Granulostasis: protein quality control of RNP granules. Front Mol Neurosci 10:84. https://doi.org/10.3389/fnmol.2017.00084
Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539(7628):197–206. https://doi.org/10.1038/nature20413
Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD (2014) Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci 34(24):8083–8097. https://doi.org/10.1523/JNEUROSCI.0543-14.2014
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495(7442):467–473. https://doi.org/10.1038/nature11922
Baron DM, Kaushansky LJ, Ward CL, Sama RRK, Chian RJ, Boggio KJ, Quaresma AJC, Nickerson JA, Bosco DA (2013) Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener. https://doi.org/10.1186/1750-1326-8-30
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GYR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Zuchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R (2017) TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95(4):808. https://doi.org/10.1016/j.neuron.2017.07.025
McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20(7):1400–1410. https://doi.org/10.1093/hmg/ddr021
Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S (2017) An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J 36(12):1669–1687. https://doi.org/10.15252/embj.201695957
Wang T, Xu W, Qin M, Yang Y, Bao P, Shen F, Zhang Z, Xu J (2016) Pathogenic mutations in the valosin-containing protein/p97(VCP) N-domain inhibit the SUMOylation of VCP and lead to impaired stress response. J Biol Chem 291(27):14373–14384. https://doi.org/10.1074/jbc.M116.729343
Nath S, Munsie LN, Truant R (2015) A huntingtin-mediated fast stress response halting endosomal trafficking is defective in Huntington’s disease. Hum Mol Genet 24(2):450–462. https://doi.org/10.1093/hmg/ddu460
Culver BP, Savas JN, Park SK, Choi JH, Zheng SQ, Zeitlin SO, Yates JR, Tanese N (2012) Proteomic analysis of wild-type and mutant Huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem 287(26):21599–21614. https://doi.org/10.1074/jbc.M112.359307
Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA (2012) Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 11(10):2006–2021. https://doi.org/10.4161/cc.20423
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12(5):1393–1407. https://doi.org/10.1091/mbc.12.5.1393
Ramdzan YM, Trubetskov MM, Ormsby AR, Newcombe EA, Sui XJ, Tobin MJ, Bongiovanni MN, Gras SL, Dewson G, Miller JML, Finkbeiner S, Moily NS, Niclis J, Parish CL, Purcell AW, Baker MJ, Wilce JA, Waris S, Stojanovski D, Bocking T, Ang CS, Ascher DB, Reid GE, Hatters DM (2017) Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep 19(5):919–927. https://doi.org/10.1016/j.celrep.2017.04.029
Hosp F, Gutierrez-Angel S, Schaefer MH, Cox J, Meissner F, Hipp MS, Hartl FU, Klein R, Dudanova I, Mann M (2017) Spatiotemporal proteomic profiling of Huntington’s disease inclusions reveals widespread loss of protein function. Cell Rep 21(8):2291–2303. https://doi.org/10.1016/j.celrep.2017.10.097
Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R (2017) The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68(4):808–820 e805. https://doi.org/10.1016/j.molcel.2017.10.015
Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P (2016) G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 212(7):845–860. https://doi.org/10.1083/jcb.201508028
Takahara T, Maeda T (2012) Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell 47(2):242–252. https://doi.org/10.1016/j.molcel.2012.05.019
Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L (2013) Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 152(4):791–805. https://doi.org/10.1016/j.cell.2013.01.033
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Goedert M (2015) NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349(6248):1255555. https://doi.org/10.1126/science.1255555
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 7(s1):S73–S87. https://doi.org/10.3233/JPD-179001
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501(7465):45–51. https://doi.org/10.1038/nature12481
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14(5):501–503. https://doi.org/10.1038/nm1746
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106(31):13010–13015. https://doi.org/10.1073/pnas.0903691106
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Investig 121(2):715–725. https://doi.org/10.1172/Jci43366
Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (2011) Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43(3):552–557. https://doi.org/10.1016/j.nbd.2011.05.001
Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, Brundin P (2012) Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One 7(6):e39465. https://doi.org/10.1371/journal.pone.0039465
Jeon I, Cicchetti F, Cisbani G, Lee S, Li ED, Bae J, Lee N, Li L, Im W, Kim M, Kim H, Oh SH, Kim TA, Ko J, Aube B, Oueslati A, Kim Y, Song J (2016) Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol 132(4):577–592. https://doi.org/10.1007/s00401-016-1582-9
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209(5):975–986. https://doi.org/10.1084/jem.20112457
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M (2013) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136(Pt 4):1128–1138. https://doi.org/10.1093/brain/awt037
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans-synaptic spread of tau pathology in vivo. PLoS One 7(2):e31302. https://doi.org/10.1371/journal.pone.0031302
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11(7):909–913. https://doi.org/10.1038/ncb1901
Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC (2000) Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 20(10):3606–3611
Hamaguchi T, Eisele YS, Varvel NH, Lamb BT, Walker LC, Jucker M (2012) The presence of Abeta seeds, and not age per se, is critical to the initiation of Abeta deposition in the brain. Acta Neuropathol 123(1):31–37. https://doi.org/10.1007/s00401-011-0912-1
Ye L, Hamaguchi T, Fritschi SK, Eisele YS, Obermuller U, Jucker M, Walker LC (2015) Progression of seed-induced abeta deposition within the limbic connectome. Brain Pathol 25(6):743–752. https://doi.org/10.1111/bpa.12252
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338(6109):949–953. https://doi.org/10.1126/science.1227157
Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M (2014) Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun 2:88. https://doi.org/10.1186/s40478-014-0088-8
Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM (2015) Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol 130(3):349–362. https://doi.org/10.1007/s00401-015-1458-4
Siman R, Lin YG, Malthankar-Phatak G, Dong Y (2013) A rapid gene delivery-based mouse model for early-stage Alzheimer disease-type tauopathy. J Neuropathol Exp Neurol 72(11):1062–1071. https://doi.org/10.1097/NEN.0000000000000006
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18(11):1584–1593. https://doi.org/10.1038/nn.4132
Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N, Lachaud C, Carrier S, Dufour N, Auregan G, Winderickx J, Hantraye P, Deglon N, Colin M, Buee L (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun 2:14. https://doi.org/10.1186/2051-5960-2-14
Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L (2010) Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal–hippocampal network. Neuron 68(3):428–441. https://doi.org/10.1016/j.neuron.2010.10.020
Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu GQ, Masliah E, Mucke L (2012) Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7(9):e45881. https://doi.org/10.1371/journal.pone.0045881
Helwig M, Klinkenberg M, Rusconi R, Musgrove RE, Majbour NK, El-Agnaf OM, Ulusoy A, Di Monte DA (2016) Brain propagation of transduced alpha-synuclein involves non-fibrillar protein species and is enhanced in alpha-synuclein null mice. Brain 139(Pt 3):856–870. https://doi.org/10.1093/brain/awv376
Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI (2014) Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA 111(29):10732–10737. https://doi.org/10.1073/pnas.1321785111
Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213(9):1759–1778. https://doi.org/10.1084/jem.20160368
Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR (2015) Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 6:6768. https://doi.org/10.1038/ncomms7768
Pearce MM (2017) Prion-like transmission of pathogenic protein aggregates in genetic models of neurodegenerative disease. Curr Opin Genet Dev 44:149–155. https://doi.org/10.1016/j.gde.2017.03.011
Babcock DT, Ganetzky B (2015) Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc Natl Acad Sci USA 112(39):E5427–5433. https://doi.org/10.1073/pnas.1516217112
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128(6):805–820. https://doi.org/10.1007/s00401-014-1343-6
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522(7556):340–344. https://doi.org/10.1038/nature14547
Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, Tolnay M (2014) Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 127(2):299–301. https://doi.org/10.1007/s00401-013-1231-5
Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M (2010) Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 330(6006):980–982. https://doi.org/10.1126/science.1194516
Eisele YS, Fritschi SK, Hamaguchi T, Obermuller U, Fuger P, Skodras A, Schafer C, Odenthal J, Heikenwalder M, Staufenbiel M, Jucker M (2014) Multiple factors contribute to the peripheral induction of cerebral beta-amyloidosis. J Neurosci 34(31):10264–10273. https://doi.org/10.1523/JNEUROSCI.1608-14.2014
Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ (2011) Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69(4):337–342. https://doi.org/10.1016/j.neures.2010.12.020
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25(25):6016–6024. https://doi.org/10.1523/JNEUROSCI.0692-05.2005
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30(20):6838–6851. https://doi.org/10.1523/Jneurosci.5699-09.2010
Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, Mollenhauer B, Schneider A (2016) Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139(Pt 2):481–494. https://doi.org/10.1093/brain/awv346
Loov C, Scherzer CR, Hyman BT, Breakefield XO, Ingelsson M (2016) alpha-Synuclein in extracellular vesicles: functional implications and diagnostic opportunities. Cell Mol Neurobiol 36(3):437–448. https://doi.org/10.1007/s10571-015-0317-0
Chistiakov DA, Chistiakov AA (2017) alpha-Synuclein-carrying extracellular vesicles in Parkinson’s disease: deadly transmitters. Acta Neurol Belg 117(1):43–51. https://doi.org/10.1007/s13760-016-0679-1
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659. https://doi.org/10.1038/ncb1596
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K (2006) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA 103(30):11172–11177. https://doi.org/10.1073/pnas.0603838103
Polanco JC, Scicluna BJ, Hill AF, Gotz J (2016) Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem 291(24):12445–12466. https://doi.org/10.1074/jbc.M115.709485
Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287(6):3842–3849. https://doi.org/10.1074/jbc.M111.277061
Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E (2014) Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging 35(8):1792–1800. https://doi.org/10.1016/j.neurobiolaging.2014.02.012
Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C (2016) Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J 35(19):2120–2138. https://doi.org/10.15252/embj.201593411
McCoy-Simandle K, Hanna SJ, Cox D (2016) Exosomes and nanotubes: control of immune cell communication. Int J Biochem Cell Biol 71:44–54. https://doi.org/10.1016/j.biocel.2015.12.006
Dieriks BV, Park TI, Fourie C, Faull RL, Dragunow M, Curtis MA (2017) alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci Rep 7:42984. https://doi.org/10.1038/srep42984
Hallett PJ, Cooper O, Sadi D, Robertson H, Mendez I, Isacson O (2014) Long-term health of dopaminergic neuron transplants in Parkinson’s disease patients. Cell Rep 7(6):1755–1761. https://doi.org/10.1016/j.celrep.2014.05.027
Mendez I, Vinuela A, Astradsson A, Mukhida K, Hallett P, Robertson H, Tierney T, Holness R, Dagher A, Trojanowski JQ, Isacson O (2008) Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat Med 14(5):507–509. https://doi.org/10.1038/nm1752
Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol 70(4):462–468. https://doi.org/10.1001/jamaneurol.2013.1933
Beekes M, Thomzig A, Schulz-Schaeffer WJ, Burger R (2014) Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol 128(4):463–476. https://doi.org/10.1007/s00401-014-1324-9
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18(2):101–113. https://doi.org/10.1038/nrn.2016.178
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71(1):35–48. https://doi.org/10.1016/j.neuron.2011.06.031
Roselli F, Caroni P (2015) From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85(5):901–910. https://doi.org/10.1016/j.neuron.2014.12.063
Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT (2012) Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb Perspect Med 2(7):a009290. https://doi.org/10.1101/cshperspect.a009290
Hardy J, Rogaeva E (2014) Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol 262 Pt B:75–83. https://doi.org/10.1016/j.expneurol.2013.11.006
Xue YC, Feuer R, Cashman N, Luo H (2018) Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis? Front Mol Neurosci 11:63. https://doi.org/10.3389/fnmol.2018.00063
Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A (2015) Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 7(307):307ra153. https://doi.org/10.1126/scitranslmed.aac8201
Paulson HL, Shakkottai VG, Clark HB, Orr HT (2017) Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat Rev Neurosci 18(10):613–626. https://doi.org/10.1038/nrn.2017.92
Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF, Zhao X (2011) Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One 6(5):e19351. https://doi.org/10.1371/journal.pone.0019351
Merk DJ, Ohli J, Merk ND, Thatikonda V, Morrissy S, Schoof M, Schmid SN, Harrison L, Filser S, Ahlfeld J, Erkek S, Raithatha K, Andreska T, Weisshaar M, Launspach M, Neumann JE, Shakarami M, Plenker D, Marra MA, Li Y, Mungall AJ, Moore RA, Ma Y, Jones SJM, Lutz B, Ertl-Wagner B, Rossi A, Wagener R, Siebert R, Jung A, Eberhart CG, Lach B, Sendtner M, Pfister SM, Taylor MD, Chavez L, Kool M, Schuller U (2018) Opposing effects of CREBBP mutations govern the phenotype of Rubinstein-Taybi syndrome and adult SHH medulloblastoma. Dev Cell 44(6):709–724 e706. https://doi.org/10.1016/j.devcel.2018.02.012
McCampbell A, Fischbeck KH (2001) Polyglutamine and CBP: fatal attraction? Nat Med 7(5):528–530. https://doi.org/10.1038/87842
Rouaux C, Loeffler JP, Boutillier AL (2004) Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol 68(6):1157–1164. https://doi.org/10.1016/j.bcp.2004.05.035
Bradshaw NJ, Bader V, Prikulis I, Lueking A, Mullner S, Korth C (2014) Aggregation of the protein TRIOBP-1 and its potential relevance to schizophrenia. PLoS One 9(10):e111196. https://doi.org/10.1371/journal.pone.0111196
Nucifora LG, Wu YC, Lee BJ, Sha L, Margolis RL, Ross CA, Sawa A, Nucifora FC Jr (2016) A mutation in NPAS3 that segregates with schizophrenia in a small family leads to protein aggregation. Mol Neuropsychiatry 2(3):133–144. https://doi.org/10.1159/000447358
Korth C (2012) Aggregated proteins in schizophrenia and other chronic mental diseases: DISC1opathies. Prion 6(2):134–141. https://doi.org/10.4161/pri.18989
Bradshaw NJ, Yerabham ASK, Marreiros R, Zhang T, Nagel-Steger L, Korth C (2017) An unpredicted aggregation-critical region of the actin-polymerizing protein TRIOBP-1/Tara, determined by elucidation of its domain structure. J Biol Chem 292(23):9583–9598. https://doi.org/10.1074/jbc.M116.767939
Knight HM, Maclean A, Irfan M, Naeem F, Cass S, Pickard BS, Muir WJ, Blackwood DH, Ayub M (2008) Homozygosity mapping in a family presenting with schizophrenia, epilepsy and hearing impairment. Eur J Hum Genet 16(6):750–758. https://doi.org/10.1038/ejhg.2008.11
Bader V, Tomppo L, Trossbach SV, Bradshaw NJ, Prikulis I, Leliveld SR, Lin CY, Ishizuka K, Sawa A, Ramos A, Rosa I, Garcia A, Requena JR, Hipolito M, Rai N, Nwulia E, Henning U, Ferrea S, Luckhaus C, Ekelund J, Veijola J, Jarvelin MR, Hennah W, Korth C (2012) Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum Mol Genet 21(20):4406–4418. https://doi.org/10.1093/hmg/dds273
Joober R, Benkelfat C, Jannatipour M, Turecki G, Lal S, Mandel JL, Bloom D, Lalonde P, Lopes-Cendes I, Fortin D, Rouleau G (1999) Polyglutamine-containing proteins in schizophrenia. Mol Psychiatry 4(1):53–57
Tanaka M, Ishizuka K, Nekooki-Machida Y, Endo R, Takashima N, Sasaki H, Komi Y, Gathercole A, Huston E, Ishii K, Hui KK, Kurosawa M, Kim SH, Nukina N, Takimoto E, Houslay MD, Sawa A (2017) Aggregation of scaffolding protein DISC1 dysregulates phosphodiesterase 4 in Huntington’s disease. J Clin Investig 127(4):1438–1450. https://doi.org/10.1172/JCI85594
Burghaus L, Eggers C, Timmermann L, Fink GR, Diederich NJ (2012) Hallucinations in neurodegenerative diseases. CNS Neurosci Ther 18(2):149–159. https://doi.org/10.1111/j.1755-5949.2011.00247.x
Baquero M, Martin N (2015) Depressive symptoms in neurodegenerative diseases. World J Clin Cases 3(8):682–693. https://doi.org/10.12998/wjcc.v3.i8.682
Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6(4):37. https://doi.org/10.1186/alzrt269
Mullard A (2018) Pfizer exits neuroscience. Nat Rev Drug Discov 17(2):86–86. https://doi.org/10.1038/nrd.2018.16
Carter PJ, Lazar GA (2018) Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov 17(3):197–223. https://doi.org/10.1038/nrd.2017.227
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537(7618):50–56. https://doi.org/10.1038/nature19323
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Hoffmann VP, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, Siemers E (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378(4):321–330. https://doi.org/10.1056/NEJMoa1705971
Kayed R, Lasagna-Reeves CA (2013) Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis 33(Suppl 1):S67–78. https://doi.org/10.3233/JAD-2012-129001
Kingwell K (2017) Zeroing in on neurodegenerative alpha-synuclein. Nat Rev Drug Discov 16(6):371–373. https://doi.org/10.1038/nrd.2017.95
Funding
This study was supported by the DGIST R&D program and Basic Science Research Program, through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (18-BD-0402, 18-BT-02, 18-01-HRSS-02, 2018R1A2B6001607 to S.B.L., and 2017R1A2B4003351, 2016M3C7A1947307, 2016M3C7A1904148 to H.L.), the Development of Platform Technology for Innovative Medical Measurements Program from the Korea Research Institute of Standards and Science (KRISS-2018-GP2018-0018) (S.B.L.), and KBRI basic research program through Korea Brain Research Institute funded by Ministry of Science and ICT (18-BR-04-03 to H.L.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts of interest among all the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chung, C.G., Lee, H. & Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 75, 3159–3180 (2018). https://doi.org/10.1007/s00018-018-2854-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2854-4