
Abstract
Bone is a dynamic tissue that depends on the intricate relationship between protein tyrosine kinases (PTK) and protein tyrosine phosphatases (PTP) for maintaining homeostasis. PTKs and PTPs act like molecular on and off switches and help modulate differentiation and the attachment of osteoclasts to bone matrix regulating bone resorption. The protein T cell ubiquitin ligand-2 (TULA-2), which is abundantly expressed in osteoclasts, is a novel histidine phosphatase. Our results show that of the two family members, only TULA-2 is expressed in osteoclasts and that its expression is sustained throughout the course of osteoclast differentiation, suggesting that TULA-2 may play a role during early as well late stages of osteoclast differentiation. Skeletal analysis of mice that do not express TULA or TULA-2 proteins (DKO mice) revealed that there was a decrease in bone volume due to increased osteoclast numbers and function. Furthermore, in vitro experiments indicated that bone marrow precursor cells from DKO mice have an increased potential to form osteoclasts. At the molecular level, the absence of TULA-2 in osteoclasts results in increased Syk phosphorylation at the Y352 and Y525/526 residues and activation of phospholipase C gamma 2 (PLCγ2) upon engagement of immune-receptor-tyrosine-based-activation-motif (ITAM)—mediated signaling. Furthermore, expression of a phosphatase-dead TULA-2 leads to increased osteoclast function. Taken together, these results suggest that TULA-2 negatively regulates osteoclast differentiation and function.
Similar content being viewed by others


Introduction
Osteoclasts are large multinucleated cells whose major function in the body is to resorb bone under physiological and pathological conditions. These cells are of monocyte-macrophage lineage, and require macrophage colony stimulating factor (M-CSF) and receptor activator of NFκB ligand (RANKL), factors produced by bone marrow stromal cells for their differentiation and maturation [1]. Reversible phosphorylation of tyrosine residues is a central regulator of cellular functions and is controlled by the opposing actions of tyrosine kinases and tyrosine phosphatases [2]. Many signaling pathways in osteoclasts depend crucially on tyrosine phosphorylation. Defective signaling by specific tyrosine kinases is implicated in several osteopetrotic mice, e.g., c-Src−/−, Pyk2−/− mice [3, 4]. The normal function of these signaling mechanisms requires regulated tyrosine phosphorylation of effector proteins, both in terms of time and amplitude, by dephosphorylation catalyzed by protein tyrosine phosphatases (PTPs). Consequently, dysregulation of PTPs produces skeletal abnormalities, as discussed briefly below.
Osteoclasts express several different types of tyrosine phosphatases [5]. It has been shown that mice carrying the moth-eaten mutation that inactivates the Src homology 2 (SH2) domain-containing tyrosine phosphatase 1 (SHP-1) have reduced bone mass due to the increased number and hyperactivity of osteoclasts [6]. This suggests that this PTP is a negative regulator of osteoclastogenesis and possibly osteoclast bone resorbing activity. PTP-PEST is a non-receptor phosphatase that is also expressed by osteoclasts, and inhibition of PTP-PEST with RNAi reduces pit formation indicating it has a positive role in osteoclast function [7]. Mice lacking PTPe exhibit a mild bone phenotype suggesting compensatory activity by other family members that have been reported in osteoclasts [8]. In general, however, the role of phosphatases in bone biology is not well known [5].
The TULA/UBASH3/STS family members, TULA/UBASH3A/STS-2 and TULA-2/UBASH3B/p70/STS-1 encoded on different chromosomes, were discovered a few years ago by several groups, including ours [9–12]. Notably, TULA-family proteins belong to the superfamily of histidine phosphatases, sharing a conserved catalytic core centered on a reactive histidine residue [13]. T cell ubiquitin ligand proteins clearly differ from classical cysteine PTPs, such as SHP-1, which is involved in the regulation of osteoclasts. TULA-family proteins exhibit a unique architecture, featuring the ubiquitin-associated (UBA), Src-homology 3 (SH3), and phosphatase domains. In spite of a substantial homology (~60 % of identity + similarity), TULA-1 and TULA-2 are quite different [14–16]. First, TULA-2 is ubiquitously expressed in mammalian cells [9, 10], whereas TULA-1 is expressed mostly in lymphocytes [10, 11] and possibly, in mast cells [17]. Second, TULA-2 is an active PTP, while the phosphatase activity of TULA-1 is drastically lower [18–20].
It has previously been shown that the lack of TULA-family proteins renders T lymphocytes hyper-reactive [10]. This finding indicates an immunomodulatory effect of TULA-family proteins. Protein tyrosine phosphatases activity of TULA-2 is essential for this effect [20]. Although the mechanism by which TULA modulates T cell reactivity is less clear, it may act as a PTP or through other mechanisms [11, 21, 22]. Protein tyrosine phosphatases activity is also essential for the regulatory role TULA-2 plays in platelets, in which the lack of TULA-2 facilitates activation in response to signaling induced through the GPVI receptor for collagen [23].
In this report, we set out to determine the role of TULA-2, a novel phosphatase in osteoclast differentiation and function. Our results show that the absence of TULA proteins in mice results in decreased bone volume due increased osteoclast numbers and function. We also demonstrate that TULA-2 regulates Syk dephosphorylation in osteoclasts and that the absence of TULA-2 in osteoclasts results in increased osteoclast bone resorption possibly through increased Syk phosphorylation and Syk-mediated signaling events.
Materials and methods
Mice
Generation of mice deficient in both TULA and TULA-2 (DKO) are previously described [10]. Mice were obtained from Dr. Nick Carpino, SUNY NY. Both DKO and the counterpart wild-type mice were maintained on a mixed C57BL/6JX129SvJ background. All mice-related experiments were performed in compliance with the Institutional Animal Care and Use Committee at University of Connecticut Health Center.
Materials
M-CSF and RANKL were purchased from R&D systems (Minneapolis, MN). Bacterial collagenase and dispase were purchased from Calbiochem (San Diego, CA). Anti-TULA-2 antibodies were raised in rabbits against synthetic peptide corresponding to either a N- or C-terminal sequence of human TULA-2: REELYSKVTPRRNRQQRPGT or GPTGGFNWRETLLQE, respectively. (The N-terminal peptide starts at the residue four of TULA-2.) It has been confirmed that both mouse and human TULA-2 react with both antibodies; this is consistent with the identity (C-terminal) or near identity (90 %) of the corresponding sequences for mouse and human TULA-2 are identical. Each antibody was affinity purified on the corresponding antigenic peptide. Both antibodies were used for Western-blot analysis. Antibody against phospho-tyrosine 4G10 was purchased from Millipore. Antibodies to phosphoTyr525/526Syk, phosphoTyr352Syk, phosphoTyr1217PLCγ2, PLCγ2 and GAPDH were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against Syk and 1,25-dihydroxyvitamin D3 and prostaglandin E2 and the leukocyte acid phosphatase kit for tartrate-resistant acid phosphatase (TRAP), reagents required for differentiation and characterization of osteoclasts were obtained from Sigma. Collagen gel was obtained from Nitta Gelatin Co., Osaka, Japan. Osteo Assay tissue culture plates for pit forming assay were purchased from Corning Life Sciences.
Skeletal analysis
High-resolution microcomputed tomography (microCT) was performed as described previously [24]. Briefly, images of long bones from 8-week-old male mice were acquired using a Skyscan 1172, 12 MPix model (Microphotonics, Allentown, PA). Proximal tibiae and distal femora were scanned with a source voltage of 59 kV, a source current of 167, an image pixel size of approximately 5.75 μm, and a 0.5-mm aluminum filter. Imaging started at the distal end of the femur or the proximal end of the tibiae and included approximately 7 mm (1,335 slices) of the total bone length. Using the CTAn software, trabecular bone was separated from cortical bone with a region of interest tool. Trabecular morphometric traits were computed from binarized images using direct 3D techniques that do not rely on prior assumptions from the underlying structures. The volume of interest for trabecular microarchitectural variables was bounded to the endocortical margin, starting 1.5 mm from the proximal tibial condyles in the direction of the metaphysis, and then extending from this position for 250 slices (1.5 mm). Upper and lower thresholds of 255 and 80 were used to delineate each pixel as ‘‘bone’’ or ‘‘non-bone,’’ and trabecular bone volume per total volume (BV/TV), mean trabecular thickness (Tb.Th.), mean trabecular number (Tb.N.), and mean trabecular separation (Tb.Sp.) indices were computed.
For histological and histomorphometric analysis, 12-week-old mice were used. To measure dynamic bone formation parameters, mice were injected with calcein (30 mg/kg body weight) 10 and 3 days before sacrifice. Tibiae and femora were dissected and fixed in 3.7 % formaldehyde in phosphate-buffered saline, preserved in 70 % ethanol, and embedded in methyl methacrylate resin. Sections (5 μm) were deplasticized and stained with von Kossa or were left unstained for the measurement of calcein labeling. Some sections were processed for TRAP staining as per manufacturer’s instructions (Sigma). For histomorphometric analysis, to assess changes in bone structure and remodeling, tibial sections were measured in the proximal metaphysis beginning 340 μm below the chondro-osseous junction of the secondary spongiosa using image analysis software (BIOQUANT Osteo II, Bioquant Image Analysis Corp., Nashville, TN) as described by Parfitt et al. [25]. Osteoblast numbers per bone surface (Ob.N./B.S.) on trabecular surfaces was determined in toluidine blue-stained sections. Osteoclast numbers per bone surface (Oc.N./B.S.; TRAP+ cells) and osteoclast surfaces on bone surfaces (Oc.S./B.S.) were determined on trabecular surfaces in the secondary spongiosa of TRAP-stained sections. Bone formation rate was calculated from calcein labeled sections.
Determination of serum collagen telopeptide
Serum was prepared from blood collected by cardiac puncture. Concentrations of C-telopeptide (CTX), a degradation product of type I collagen, in serum of 12-week-old mice were determined using the Rat Laps ELISA (Osteometer BioTech A/S, Herlev, Denmark).
Adenoviral constructs
Adenovirus vectors carrying the reporter gene encoding green fluorescent protein (GFP; AxGFP) and the FLAG-tagged TULA-2 phosphatase-dead mutant (AxTULA-2H391A) mutant were generated, as previously described [26, 27].
Analysis of osteoclast precursors by fluorescence activated cell-sorter
Antibodies used for flow cytometric analysis were as the following: anti-mouse CD3 APCe780, anti-mouse B220 APCe780, anti-mouse CD11b FITC. Anti-mouse CD115-biotin was used in combination with streptavidin–phycoerythrin (PE). All antibodies were purchased from eBiosciences (San Diego, CA). To obtain cells, tibiae and femora of 8-week-old mice were flushed with staining medium (1× Hanks balanced salt solution (HBSS), 10 mM HEPES, 2 % newborn calf serum) using 25-gauge needle. After washing cells, red blood cells were lysed with ammonium chloride and cells were filtered through a 40-μm cell strainer to remove cellular debris. Cells were incubated with primary antibodies for 45 min on ice. After washing, cells were incubated with secondary antibodies, when necessary, for 45 min time on ice. Cells were washed and resuspended in staining medium containing propidium iodide (1 μg/ml) and populations of interest were analyzed by using LSRII flow cytometer (BD Biosciences) excluding the dead cells. For some experiments cells were first gated on CD45R, CD3, and NK1.1 negative (Triple Negative) population and then separated as CD11b−/low CD115+ population. To measure LSK (Sca1+, c-Kit+ Lin+) population, cells were labeled with biotinylated mouse lineage panel antibodies (BD Bioscience) followed by PerCP-conjugated streptavidin and stem cell markers Sca-1 (FITC-conjugated) and c-Kit (APCe780-conjugated). A lineage negative (Lin-) population was gated for further analysis of stem cell phenotypes (Sca1+, c-Kit+ Lin−).
Colony-forming unit assay
Colony-forming assays were performed, as previously described [28]. Briefly, bone marrow cells (1 × 105 cells) were cultured in α-MEM containing 3 % methylcellulose, 20 % FBS, 1 % bovine serum albumin (BSA) and recombinant murine GM-CSF (1 ng/ml). Cells were plated in volume of 1 ml in 35-mm culture dishes and incubated at 37 °C in a humidified atmosphere of 5 % CO2-air for 7 days. Colonies composed of 50 or more cells were counted after 7 days using an inverted microscope.
Cell culture
For generation of osteoclast-like cells (OCLs), bone marrow was isolated from tibia and femur of 4 to 6-week-old mice. Following overnight incubation, the non-adherent cells were plated at 2.5 × 105/cm2 in α-MEM medium containing 10 % FBS and 20 ng/ml M-CSF. Subsequently, cells were treated with M-CSF (20 ng/ml) and RANKL (50 ng/ml) for additional 5–6 days. For some experiments, OCLs were also generated by the co-culture method as previously described [29, 30]. Briefly, mouse primary osteoblastic cells were obtained from 1-day-old mouse calvaria by enzymatic digestion. Bone marrow cells (105 cells/cm2) were co-cultured with calvarial cells (2 × 104 cells/cm2) on tissue culture plates or collagen gel-coated plates in the presence of 10 nM 1,25-dihydroxy vitamin D3 and 1 μM prostaglandin E2 (Sigma). For some experiments, osteoclasts were infected with adenoviruses bearing TULA-2 mutants as described previously [29, 30].
For the generation of the bone marrow macrophages (BMMs), cells isolated from long bones were plated onto polystyrene culture plates and cultured α-MEM containing 10 % FBS, and 50 ng/ml M-CSF for 72 h. Cells were then trypsinized, counted and plated onto six-well culture plate (106 cells/well).
RAW 264.7 cells were cultured in Dulbecco’s modified essential medium supplemented with 10 % FBS. Cell lysates were prepared as described below.
RT-PCR analysis
The expression levels of osteoclast differentiation and fusion markers were analyzed by quantitative real-time PCR. Sequences for the primers and details of the procedure are described previously [31].
Pit formation assay
Functionally active OCLs were formed in co-cultures, as described above. An aliquot of the crude OCL preparation was transferred onto hydroxyapatite-coated dishes and cultured for an additional 48 h. The resorbed area was measured using an image analysis system linked to a light microscope (Bioquant Osteo II). A similar aliquot was cultured onto a 24-well dish and cells were TRAP stained after 6 h.
Survival assay
Following differentiation with MCSF and RANKL on day 5, one set of 96-well plate was fixed with 10 % formaldehyde in PBS. Other sets were either kept in α-MEM or were treated with RANKL (50 ng/ml) for 24 h. Cells were fixed and TRAP stained using a commercial kit (Sigma, St. Louis). Total number of TRAP + multinucleated cells were counted and expressed as percentage of the number of cells at the start of the experiment on day 5.
Preparation of cell lysates and immunoprecipitation
For M-CSF and RANKL stimulation, cells were serum starved for 1 h and stimulated with either M-CSF (50 ng/ml) or RANKL (50 ng/ml) for the indicated time period. For Fc receptor stimulation, cells were washed once with serum-free medium and incubated with 10 μg/ml mouse IgG primary antibody (Jackson ImmunoResearch, West Grove, PA) for 30 min at 4 °C. Cells were washed once with serum-free medium and incubated for additional 15 min at 37 °C. Cells were then stimulated by crosslinking with goat anti-mouse secondary antibody (Jackson ImmunoResearch) for the indicated time period at 37 °C. Stimulation of cells was terminated by washing the cells once with ice-cold PBS and flash freezing with liquid N2. Flash-frozen samples were thawed on ice and lysed in lysis buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 0.05 % Nonidet P-40, 10 % glycerol, 10 mM EDTA, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride. After 30 min on ice, lysates were cleared by centrifugation at 12,000×g for 20 min. Lysates were then used for analysis of proteins by Western blotting as previously described. For immunoprecipitation, lysate (750 μg) was incubated with primary antibody (1 μg) for 30 min on ice. Washed Sepharose beads were resuspended in PBS and at a ratio of 1:5 and 40 μl of the suspended beads were added to the lysate/antibody mixture and incubated at 4 °C for 1 h on end-on-end rocking shaker. Beads were then washed three times in the above lysis buffer. Proteins bound to beads were eluted by incubating beads with equal volume of 10 mM glycine (pH 3.0) at room temperature for 30 min. Eluted proteins were mixed with equal volume of SDS loading buffer and boiled for 5 min and loaded on to SDS-PAGE gel.
Statistics
Each experiment was repeated at least three times, unless indicated otherwise. The results obtained from a typical experiment were expressed as the mean ± standard deviation. Statistical analysis was performed using GraphPad Prism 4. Significant differences were determined using students t test; p < 0.05 was considered significant.
Results
TULA-2 is the only family member expressed in osteoclasts precursors and mature osteoclasts
Previous reports indicated that in contrast to TULA, which exhibits expression restricted to certain cell types, TULA-2 is ubiquitously expressed [14–16]. We examined the expression pattern of TULA-2 mRNA during osteoclast differentiation and compared it to that of PU.1, RANK and Calcitonin receptor (CalcR), proteins differentially expressed in the process of osteoclast maturation. As shown in Fig. 1a, TULA-2 is expressed already in osteoclast precursors, and, based on an increase in the intensity of the amplified bands, its expression appears to increase after addition of M-CSF or M-CSF + RANKL. In contrast, expression of TULA was undetectable during osteoclastogenesis corroborating previous reports that TULA expression is restricted [10]. These results were confirmed using Western-blot analysis, which clearly revealed the presence of TULA-2 in the precursors and its increase in the course of differentiation (Fig. 1b) and the lack of detectable TULA (data not shown). Together, these results indicate that of the TULA-family of proteins, only TULA-2 is expressed in osteoclast precursors and mature osteoclasts.

Expression profile of TULA-2 during osteoclast differentiation. a Bone marrow precursors were cultured in the presence of macrophage colony stimulating factor (M-CSF)alone or with M-CSF and receptor activator of NFκB ligand (RANKL), for the indicated number of hours. Expression levels of PU.1, RANK, Calcitonin receptor, TULA-2, TULA and GAPDH were determined by using specific primers. Five microliters of sample was loaded onto an agarose gel to visualize bands. cDNA derived from primary T cells was used as a positive control (C). b Expression of TULA-2 protein was determined in precursor and mature osteoclast lysates (OCLs). Cells were treated with cytokines for indicated hours. Total cell lysates were electrophoresed and Western blotted with anti-TULA-2 antibodies (top panel). Cell lysate derived from macrophage cell line RAW297.4 was used as a positive control (C). Blot was re-probed with anti-actin antibodies (middle panel) to determine protein loading. Prior to addition of antibodies blot was also stained with Ponceau stain (lower panel) to detect protein transfer onto the nitrocellulose paper
Next, in order to determine the intrinsic effects of the loss of TULA-2 during osteoclast differentiation, bone marrow cells from mice deficient in TULA and TULA-2 (DKO) were used. Bone marrow precursors were differentiated into mature osteoclasts in the presence of M-CSF and RANKL. Following TRAP staining, the total number of TRAP positive cells and the number of osteoclasts with more than 20 nuclei were counted. As indicated in Fig. 2a and b, both total numbers of cells and cells with 20+ nuclei were significantly increased in DKO cultures. To test if the observed increase in osteoclasts in the DKO cultures is due to enhanced differentiation or fusion of osteoclast precursor cells, expression of osteoclast differentiation markers was examined by real-time PCR analysis. No significant differences were found between the expression levels of PU.1, RANK, Calcitonin Receptor and TRAP, which are used as differentiation markers (Fig. 2d–g). Similarly, expression of DC-STAMP and OC-STAMP, fusion markers, was comparable between the WT and DKO cells (Fig. 2h, i).

Absence of TULA proteins results in increased osteoclast numbers in ex vivo cultures. a Non-adherent bone marrow precursors were cultured in the presence of M-CSF for 2 days and for additional 3 days in the presence of M-CSF and RANKL. TRAP staining showed increased in numbers of TRAP+ cells in DKO cultures on day 5. Photomicrographs show TRAP-stained OCLs at 4× (upper panels) and 20× (lower panels) magnification. b The numbers of TRAP+ multinucleated osteoclasts (MNCs) in the DKO cultures (black bars) were significantly greater than in WT cultures (white bars) at day 5. c Cells with more than 20–50 nuclei were increased in numbers DKO cultures. d–i. Bone marrow cells were cultured in the presence of MCSF (20 μg/ml) and RANKL (50 μg/ml) for 5 days and expression of the following osteoclast differentiation markers d PU.1; e RANK; f CalcR; and g TRAP; and fusion markers h DC-STAMP and i OC-STAMP was determined by real-time PCRs
An increase in osteoclast numbers could be due to enhanced ability of cells to survive. Therefore, we next examined the ability of mature osteoclasts to survive in cultures in response to MCSF or RANKL. Two sets of osteoclast cultures were derived from bone marrow, as described above. Upon the appearance of large multinucleated cells, one set was fixed and the second set was incubated in serum-free media supplemented MCSF or RANKL for an additional 24 h. Cells were then TRAP-stained and the percent survival was calculated by normalizing the number of TRAP-stained osteoclasts counted after the additional 24-h incubation period to the number of osteoclasts fixed immediately after the appearance of osteoclasts. Comparable survival was observed between WT and DKO cultures under all conditions (Supplementary Figure 1).
Numbers of osteoclast precursors in the bone marrow are increased in the absence of TULA proteins
Osteoclasts are of hematopoietic origin and share the same lineage as macrophages. It has been reported that bone marrow (BM) cells that negative for markers of B-lymphocytes (CD45R, also known as B220), and macrophages (CD11b), but positive for M-CSF receptor c-Fms (CD115), are osteoclast precursors [32, 33]. These populations are distinct from hematopoietic stem cells because of their lack of reactivity to Sca-1 antibody. This population represents less than 2 % of the fresh BM preparations and has the highest levels of in vitro osteoclastogenic activity [33]. Our FACS analysis results comparing the WT and DKO BM cells demonstrated that the numbers of CD11b+ cells were comparable, while the number of CD45−CD3−CD11b−/low CD115+ cells were doubled in the DKO mice, as compared to the WT mice (Fig. 3a–d). The granulocyte macrophage progenitor (CFU-GM) is the earliest identifiable osteoclast precursor and can become osteoclasts in addition to macrophages or granulocytes [28]. Colony-forming unit assays showed that in the presence of GM-CSF DKO BM cells formed twice as many colonies than WT cells (Fig. 3e), suggesting that DKO mice have more osteoclast precursors.

Numbers of osteoclast precursors are increased in DKO mice. Bone marrow cells obtained from long bones were analyzed for the expression of CD11b and CD115 by FACS analysis. Dot plots demonstrating gating parameters and frequencies of different subpopulations for a representative WT (a) and DKO (b) mouse is shown. Numbers represent percentages of parent population. c Bar graphs represents percentage of total live cells in bone marrow for CD45R−, CD3−, NK1.1− (TN), CD11b+ cells. d Bar graphs represents percentage of total live cells in bone marrow CD45R−, CD3−, NK1.1− (TN), CD11b−/low CD115+. n = 3 mice/group, mean ± SD; **p < 0.0001 compared to WT. e BM cells (1 × 105 cells/culture) were prepared for colony-forming assays as described in methods. The cultures were incubated in humidified chamber for 7 days. Clusters of 50 or more cells were scored as a colony. Bar graph shows the number of colonies formed. n = 7 samples, mean ± SD shown; **p < 0.0001 compared to WT. The experiment was repeated twice
DKO mice have decreased bone volume due to increased numbers and functions of osteoclasts
Gross radiological analysis of long bones from 12-week-old DKO mice showed decreased bone density in the femur and tibia as compared to the age-matched control WT mice (Supplementary Figure 2). MicroCT analysis of long bones of the hind limb indicate that bone volume was significantly decreased in the DKO mice (Fig. 4). In the DKO long bones, although trabecular separation was not affected, a significant decrease in trabecular thickness and number was observed, compared to WT samples (Fig. 4b, d). Histological examination of von Kossa-stained sagittal sections of the proximal tibia from age-matched WT and DKO mice indicated a significant decrease in mineralized trabecular bone in DKO mice as compared to WT (Fig. 5a). This result was confirmed with histomorphometry of von Kossa-stained bone sections (data not shown). Also, no differences were observed in cortical thickness (data not shown). A decrease in bone volume can result from either a decrease in bone formation by osteoblasts or an increase in bone resorption by osteoclasts. Therefore, in order to determine which cell type contributed to the decreased bone volume in DKO mice, we compared the number and activity of both osteoblasts and osteoclasts in DKO and WT mice. Toluidine blue staining of the long bones indicated that the numbers of osteoblasts were comparable in WT and DKO samples (Fig. 5c). Similarly, bone formation rate as measured by calcein double labeling was also comparable between the WT and DKO mice (Fig. 5d). On the other hand, quantification of TRAP-stained cells in the cancellous bone indicated that there was a twofold increase in osteoclast numbers and osteoclast surface/bone surface, in DKO mice as compared to WT (Fig. 5b, e, f).

Absence of TULA proteins results in decreased bone volume. Micro-CT analysis of tibia (a) and femur (c) from WT and DKO mice. a and c shows representative sagittal sections of tibia and femur, respectively. Histograms show the trabecular parameters of tibia (b) and femur (d). n = 4 per genotype, 8-week-old male mice, mean ± SD shown; *p < 0.05, **p < 0.01 compared to WT

Absence of TULA proteins results in decreased bone volume due to increased osteoclast numbers. a Representative histological sections of proximal tibiae of 12-week-old WT and DKO mice stained with Von Kossa and counterstained with Toluidine blue shows decreased mineralized bone in the metaphyseal trabecular region of DKO samples. Images were taken using a 10× objective. b Tibial sections from 12-week-old WT and YF were stained for TRAP (400× magnification). Two different sections for each genotype are shown. c Quantification of number of osteoblasts/bone surface (Ob.N./BS), and d dynamic parameters of bone formation were assessed using tibias harvested from 8-week-old WT and YF mice that were injected with calcein. as described in the methods. Bone formation rate/bone surface (BFR/BS, μm2/μm/day) is shown. e Osteoclast numbers/bone surface (Oc.N./BS %), and f osteoclast surface/bone surface (Oc.S./BS). n = 4 mice per genotype; mean ± SD shown; **p < 0.01, compared to WT
We next examined the effect of absence of TULA-2 on osteoclast function. Serum levels of the C-terminal collagen telopeptide (CTX), a serum biomarker of osteoclast activity, were increased in DKO mice as compared to WT mice (Fig. 6a). To confirm this defect of DKO osteoclast function, we then examined bone resorption using in vitro pit formation assay. DKO and WT OCLs were generated by co-culture with osteoblasts on collagen gel, as described previously [29]. After 5 days in culture, a portion of the crude OCL preparation was placed on hydroxyapatite-coated tissue culture plates for an additional 48 h. The resorbed area was quantified using image analysis and normalized to the number of OCLs. DKO OCLs resorbed more surface area/cell than the WT OCLs (Fig. 6b, c), confirming the cell autonomous nature of the observed defect in osteoclast function. Thus, the decreased bone volume in the adult DKO mice and increased bone resorption in vivo and in vitro is, at least in part, due to impaired osteoclast function. Taken together, these results suggest that bone resorption under basal conditions is affected in DKO mice.

Absence of TULA-2 in osteoclasts results in increased activity both in vivo and in vitro. a Measurement of serum collagen telopeptide (CTX) demonstrated increased osteoclast activity in the DKO mice (n = 4 male mice per genotype; mean ± SD shown; **p < 0.01, compared to WT). b Photomicrographs of the resorbed area. c Bar graph demonstrating pit formation activity of DKO osteoclasts. Osteoclasts were generated by co-culture method, as described in the methods. After removing adherent cells, resorbed area was quantified and normalized for the number of osteoclasts. Data are presented as mean SE (n = 9). **p < 0.001 compared to the WT samples
Syk phosphorylation and Syk-mediated signaling is augmented in DKO cells
The increased bone resorption by osteoclasts in the absence of TULA-2 (Fig. 6) suggested that osteoclast function is modified by DKO, because signaling events are perturbed by the absence of TULA-2. Therefore, we next determined the effects of the loss of TULA-2 on response of BMMs and osteoclasts to M-CSF and RANKL, key physiological stimuli of this cell lineage (Fig. 7a, data not shown). Upon stimulation with RANKL, no significant increase in total tyrosine phosphorylation was evident in either WT or DKO osteoclasts. However, an increase in protein phosphorylation was observed both in WT and DKO cells upon treatment with M-CSF. Notably, both the M-CSF-caused increase and the basal tyrosine phosphorylation were elevated, albeit modestly, in DKO cells as compared to WT cells (Fig. 7a).

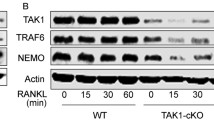
Loss of TULA-2 leads to enhanced signaling down stream of ITAM-mediated co-stimulatory pathway. Bone marrow macrophages (BMMs) were generated from the bone marrow of WT and DKO mice. a BMMs were stimulated by with M-CSF (50 ng/ml) or RANKL (50 ng/ml) at indicated time points, shown in minutes. Blots were probed with anti-phosphotyrosine antibodies (upper panels) to visualize phosphorylated proteins. Blots were then reprobed with anti-GAPDH antibodies to determine protein loading. b BMMs were stimulated by cross-linking Fc receptor to activate the signaling pathway downstream of ITAM receptors. Blot was probed with anti-phosphotyrosine antibodies (upper panel). Asterisk indicates location of proteins that are hyper-phosphorylated upon stimulation in DKO samples. Blots were reprobed with anti-GAPDH antibodies to determine protein loading. c BMMs were stimulated by cross-linking Fc receptor and immunoprecipitation with anti-PTyr was performed as described in the methods. Blot was probed with anti-PTyr antibodies (upper panel) and reprobed with anti-Syk antibodies (lower panel). The amounts of total proteins in individual bands were quantified by using Odyssey Infrared Imaging Systems software 2.1 (LICOR Biosciences). The ratio of tyrosine phosphorylation to amount of total protein is shown at the bottom of the panel. d, e BMMs were pretreated with either 50 μg/ml of the Syk inhibitor piceatannol (PICE) or DMSO (VEH) and were then stimulated to engage the ITAM receptors as described above. d Blot was probed with anti pY525/526 Syk or pY352Syk as indicated. Blots were then probed with anti-Syk antibodies to determine loading. e To determine PLCγ2 phosphorylation blots were probed with anti-pY1217PLCγ2 antibodies and then reprobed with anti-PLCγ2 antibodies to determine protein loading. The amounts of total proteins in individual bands were quantified by using Odyssey Infrared Imaging Systems software 2.1 (LICOR Biosciences). The ratio of tyrosine phosphorylation to amount of total protein is shown at the bottom of the panel
Syk is a known substrate of TULA-2 [19] and a key factor in transmitting the ITAM-mediated signaling downstream of FcγR in several cell types, including BMMs [34]. Downstream of ITAM-mediated signaling Syk is also required for osteoclast development and function [35–37]. Therefore, we next examined the phosphorylation events upon engagement of FcγR in BMMs. After Fc receptor crosslinking, there was a robust increase in total tyrosine phosphorylation, which was higher in DKO BMM than in WT BMM (Fig. 7b). Tyrosine phosphorylation of proteins migrating at approximately 75 kD and above was enhanced in DKO cells as compared to WT cells. To reduce background caused by cross-reactivity, we enriched tyrosine-phosphorylated proteins by immunoprecipitating them using anti-pTyr antibody and then immunoblotted the obtained immunoprecipitates for total tyrosine phosphorylation and Syk. An increase in tyrosine phosphorylation was observed in the DKO as compared to WT lysates. Multiple tyrosine-phosphorylated proteins were detected in BMM starting at 72 kDa and above it with approximately 10 kDa increments; the number of discernable bands is much higher in DKO cells than in WT cells (Fig. 7c). Total anti-pTyr reactivity normalized to the amount of Syk is increased by ~1.8-fold in DKO vs. WT cells (Fig. 7c).
Given that Syk has several tyrosine residues that can be differentially phosphorylated in DKO and WT cells and may exhibit differential reactivity to a total anti-pTyr antibody, we next analyzed the phosphorylation status of individual tyrosine residues of Syk using specific anti-phosphotyrosine site antibodies. For this analysis we selected Y352 and Y525/Y526; the former is the site known to preferentially dephosphorylated by TULA-2 [19], while the latter is a site of known importance for Syk function [38, 39]. As expected, Western-blot analysis showed an increase in tyrosine phosphorylation for both Y525/526 and Y352 sites in both WT and DKO cells. However, tyrosine phosphorylation in DKO cells was elevated as compared to that in WT cells by ~1.5- to 2-fold (Fig. 7d). We next examined tyrosine phosphorylation of PLCγ2, a key signaling protein involved in Syk-mediated signaling, in WT and DKO cells. Although, in the absence of stimulation, basal phosphorylation of PLCγ2 tyrosine 1,217 was higher in DKO than in WT, it was substantially increased in response to Fc receptor crosslinking (Fig. 7e).
To further elucidate the involvement of Syk in these events, we treated cells in our experiments with piceatannol, a Syk specific inhibitor [37]. Phosphorylation of the Syk sites examined did not change profoundly, while the observed increase in PLCγ2 tyrosine 1,217 phosphorylation was dramatically decreased (Fig. 7e). This suggests that phosphorylation of Syk Y352 and Syk Y525/526 in this cellular context is largely independent of Syk enzymatic activity (it can be carried out by Src-family kinases, for example), while phosphorylation of PLCγ2 is clearly Syk-dependent. Thus, differential phosphorylation of Syk caused by differential activity of TULA-2 is expected to exert a strong effect on PLCγ2 phosphorylation, which is a key event of signaling via FcRγ and other osteoclast/macrophage receptors and other cells of immune system [36, 40, 41].
TULA-2 phosphatase activity is needed for osteoclast-mediated bone resorption
TULA-2 is highly expressed in osteoclasts (Fig. 1); its absence in osteoclasts resulted in increased phosphorylation of Syk (Fig. 7d). To investigate the role of TULA-2 phosphatase activity in osteoclast function, we constructed adenoviruses containing FLAG-tagged phosphatase-dead TULA-2 (AxTULA-2H391A). We used a replication-deficient adenovirus vector that contains a reporter gene encoding GFP (AxGFP) as a control vector. Expression of TULA-2 in OCLs isolated from co-cultures infected with the recombinant viruses was verified after 3 days of infection using Western blotting with anti-FLAG and anti-TULA-2 antibodies (data not shown). To investigate the functional consequences of overexpressing phosphatase-deadTULA-2, the bone-resorbing activity of OCLs was quantified by measuring the areas of pits formed on hydroxyl apatite-coated resorption surface. The activity of OCLs phosphatase-dead TULA-2 was significantly augmented when compared to control and GFP-expressing osteoclasts (Fig. 8a, b). This indicates that TULA-2 phosphatase activity is required for bone resorption.

TULA-2 phosphatase activity is required for proper osteoclast function. Osteoclasts were either left un-infected, or were infected at 500 MOI with an adenoviral GFP (AxGFP) or adenoviral construct expressing phosphatase-dead form of TULA 2 (AxTULA-2H380A) for 48 h. Infected cells were replated onto the resorbable tissue culture surface and incubated for 48 h as described in the methods section. a Photomicrographs show of resorbed surface for each condition. b After removing adherent cells, resorbed area was quantified and normalized for the numbers of OCLs. Data are presented as mean SE (n = 9). **p < 0.001 compared to the uninfected control samples. The results are representative of two experiments
Discussion
We have shown in the present study that the absence of TULA-2, a novel PTP that belongs to the histidine phosphatase superfamily, leads to the decreased bone volume in TULA-2-deficient/null mice (Figs. 4, 5). This is apparently due to both the marked increase in the ability of the hematopoietic precursor cells to differentiate into osteoclasts (Fig. 2a, b) and the enhanced ability of osteoclasts to resorb bone both in vivo and in vitro (Fig. 6). At the molecular level, our studies show that in osteoclast precursors, TULA-2 mediates dephosphorylation of Syk, which is known to regulate osteoclast differentiation and function via ITAM signaling [36, 42].
Both members of the TULA family have been proposed to be negative regulators of signaling. Deletion of both TULA and TULA-2 is required for causing dramatic hyperactivation of T cells [10]. Re-expression of WT, but not inactivated TULA-2 in cells that lack both TULA-family members results in substantial reversal of the phenotype, indicating that TULA-2 PTP activity is responsible, at least in part, for the hyper-responsiveness of T cells [20]. In the present study, we show that only TULA-2 is expressed in developing and mature osteoclasts to a measurable extent (Fig. 1). This finding is in line with the observation that TULA has limited expression [14, 15]. Therefore, any effect seen in osteoclasts in the DKO mice can be attributable to TULA-2 deficiency.
The overall effect of the lack of TULA-2 on the bone/bone phenotype is due to increased osteoclast numbers in vivo (Fig. 5e) and in vitro (Fig. 2a–c) and increased osteoclast function (Fig. 6). We cannot, however, rule out the possibility that the absence of TULA proteins also affects the osteoblast lineage and this effect of TULA/TULA-2 deficiency on osteoblasts contributes to the osteopenic phenotype. This possibility is supported by a decrease in trabecular thickness and trabecular numbers in DKO mice, when compared to the WT mice (Fig. 4). Although further studies would be needed to elucidate how loss of TULA proteins might affect the function of osteoblasts, it is likely that this effect would be indirect, being mediated, for example, by changes in cytokine production. The finding that an increase in osteoclastogenesis in DKO mice is intrinsic to the hematopoietic lineage excludes the possibility that the observed increase is due to an increase in osteoprotegerin ligand (OPGL) or a decrease in osteoprotegerin production by cells of the stromal lineage or osteoblasts [43, 44].
Given the distinct sequential and essential effects by M-CSF on osteoclast precursor monocyte proliferation and survival, and by RANKL on osteoclast differentiation [1], we attempted to dissect the roles of these cytokines in the enhanced osteoclastogenesis that accompanies TULA-2 deficiency. A modest increase in phosphorylation was seen in the DKO osteoclast precursors or mature osteoclasts as compared to their WT counterparts stimulated with M-CSF (Fig. 7a). Furthermore, a significant increase in phosphorylation of several proteins, including Syk, was observed in DKO cells as compared to WT cells when ITAM-mediated signaling was engaged (Fig. 7b, c, e). Although the exact molecular mechanisms responsible for TULA-2-modulated regulation of osteoclastogenesis remain unresolved, we postulate that decreased TULA-2 activity elevates tyrosine phosphorylation of Syk and, hence, of its substrates, thus leading to increased osteoclastogenesis in TULA-2-deficient mice, analogous to the osteopenia observed in SHP-1-deficient mev/mev mice [6]. Under normal remodeling conditions, bone resorption is thought to stimulate an equivalent level of bone formation. Osteoclast and osteoblast “coupling”, is fine-tuned through variations in local cytokine expression or release, from cell to cell, or from cell to matrix induced signals [45]. However, our studies in DKO mice show that increased osteoclastogenesis occurs independently of osteoblastogenesis, consistent with other studies where osteoclast resorptive activity appears dispensable for coupling [46]. Thus, the increase in osteoclast levels and activity that we observed histologically, functionally, and independently of bone formation contribute to the significant loss of trabecular bone volume in DKO mice. Therefore, overall bone-remodeling rates appear to be reduced arguing further for uncoupling of osteoclast and osteoblast functions in the DKO mice.
Although the total numbers of bone marrow cells and hematopoietic progenitors (Lin−Sca-1+c-kit + cells) remained unchanged in the DKO mice, the numbers of osteoclast precursors (CD11b−/low, CD115+, CD117+) were significantly increased (Fig. 3 and data not shown). This finding is consistent with increased osteoclastogenesis wherein DKO osteoclast precursor cells differentiate into osteoclasts (Fig. 2) and have increased resorption (Fig. 6). In addition to bone formation, bone remodeling is also tightly linked to hematopoiesis. Indeed, osteoclasts appear to have a unique, direct role in hematopoietic progenitor mobilization [47]. Thus, our findings also suggest the possibility that enhanced osteoclastogenesis could have a positive feedback effect on hematopoietic progenitors and drive their differentiation toward different lineages, due in part to release of soluble factors from the osteoid matrix degraded by the increased osteoclast activity in the DKO mice. Overall, the absence of TULA proteins and, in particular TULA-2, since only TULA-2 is expressed in osteoclasts, leads to bone loss.
Taken together, these results indicate that, under normal physiological conditions TULA-2 could down-regulate signaling in response to factor(s) that activate differentiation of osteoclasts and possibly osteoclast function. Although the exact mechanisms mediating the role of TULA-2 remain to be elucidated further, the obtained results allow us to propose a molecular explanation for the positive effect of the loss of TULA-2 activity on osteoclastogenesis and bone resorption in DKO mice. Considering that the protein tyrosine kinase Syk as a key element of the signaling mechanism in macrophages and osteoclasts [36, 40, 41] and that Syk has been identified as a bona fide substrate of TULA-2 [18, 19], it is likely that the lack of TULA-2 facilitates phosphorylation of Syk. Indeed, our results indicate that phosphorylation of Syk and, in particular, phosphorylation of Tyr 525/526 and Tyr 352 sites is increased in TULA-2-deficient cells. It has been shown previously that the elevated phosphorylation of these sites is indicative of elevated enzymatic activity of Syk [34]. The hyper phosphorylation of Syk demonstrated in this report is similar to that reported for TULA-2-deficient platelets [23].
Likewise, we have shown that tyrosine phosphorylation of PLCγ2 is also elevated in the osteoclast precursors derived from DKO mice as compared to WT precursors. An increase in PLCγ2 phosphorylation in DKO cells is likely due to the increase in Syk activity caused by the lack of TULA-2, since we (Fig. 7e) and others have shown that PLCγ2 phosphorylation is Syk-dependent [23]. Direct dephosphorylation of PLCγ2 pTyr 1,217 by TULA-2 is less likely, since this site lacks some critical specificity determinants defining it as a substrate of TULA-2 [19].
In addition to Syk, Src-family kinases (SFKs) may also be targets of TULA-2 phosphatase activity. Several SFKs also regulate bone remodeling by directly affecting osteoclast differentiation and function. While absence of c-Src does not affect osteoclast differentiation, it severely affects osteoclast function, thus causing osteopetrosis in mice [4]. In contrast, while the lack of Lyn does not impact the activity of mature osteoclasts under basal condition, Lyn-null mice undergo accelerated osteoclastogenesis and bone loss in response to RANKL [48]. Similarly, while Fyn-deficient mice show no overt changes in the skeletal phenotype, the absence of Fyn results in decreased osteoclast differentiation in culture [49]. Thus, if TULA-2 regulates kinase activities of SFKs, its absence in osteoclast may result in dysregulation of SFKs and could lead to the increased numbers of osteoclasts and an increase in osteoclastic activity.
In conclusion, the absence of TULA-2 leads to osteopenia accompanied by increased bone resorption. Both osteoclast differentiation and osteoclast activity appear to be increased in the absence TULA-2, suggesting that this tyrosine phosphatase is a negative regulator of osteoclastogenesis and, possibly, of osteoclast resorbing activity.
References
Teitelbaum SL, Ross FP (2003) Genetic regulation of osteoclast development and function. Nat Rev Genet 4:638–649
Hunter T (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80:225–236
Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, Schlessinger J (2007) Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(−/−) mice. J Cell Biol 178:1053–1064
Soriano P, Montgomery C, Geske R, Bradley A (1991) Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64:693–702
Granot-Attas S, Elson A (2008) Protein tyrosine phosphatases in osteoclast differentiation, adhesion, and bone resorption. Eur J Cell Biol 87:479–490
Aoki K, DiDomenico E, Sims NA, Mukhopadhyay K, Neff L, Houghton A, Amling M, Levy JB, Horne WC, Baron R (1999) The tyrosine phosphatase SHP-1 is a negative regulator of osteoclastogenesis and osteoclast resorbing activity: increased resorption and osteopenia in me v /me v mutant mice. Bone 25:261–267
Chellaiah MA, Kuppuswamy D, Lasky L, Linder S (2007) Phosphorylation of a Wiskott–Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem 282:10104–10116
Chiusaroli R, Knobler H, Luxenburg C, Sanjay A, Granot-Attas S, Tiran Z, Miyazaki T, Harmelin A, Baron R, Elson A (2004) Tyrosine phosphatase epsilon is a positive regulator of osteoclast function in vitro and in vivo. Mol Biol Cell 15:234–244
Carpino N, Kobayashi R, Zang H, Takahashi Y, Jou ST, Feng J, Nakajima H, Ihle JN (2002) Identification, cDNA cloning, and targeted deletion of p70, a novel, ubiquitously expressed SH3 domain-containing protein. Mol Cell Biol 22:7491–7500
Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN (2004) Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 20:37–46
Feshchenko EA, Smirnova EV, Swaminathan G, Teckchandani AM, Agrawal R, Band H, Zhang X, Annan RS, Carr SA, Tsygankov AY (2004) TULA: an SH3- and UBA-containing protein that binds to c-Cbl and ubiquitin. Oncogene 23:4690–4706
Wattenhofer M, Shibuya K, Kudoh J, Lyle R, Michaud J, Rossier C, Kawasaki K, Asakawa S, Minoshima S, Berry A, Bonne-Tamir B, Shimizu N, Antonarakis SE, Scott HS (2001) Isolation and characterization of the UBASH3A gene on 21q22.3 encoding a potential nuclear protein with a novel combination of domains. Hum Genet 108:140–147
Rigden DJ (2008) The histidine phosphatase superfamily: structure and function. Biochem J 409:333–348
Tsygankov AY (2008) Multidomain STS/TULA proteins are novel cellular regulators. IUBMB Life 60:224–231
Tsygankov AY (2009) TULA-family proteins: an odd couple. Cell Mol Life Sci 66:2949–2952
Tsygankov AY (2012) TULA-family proteins: a new class of cellular regulators. J Cell Physiol 228(1):43–49
de Castro RO, Zhang J, Groves JR, Barbu EA, Siraganian RP (2012) Once phosphorylated, tyrosines in carboxyl terminus of protein-tyrosine kinase Syk interact with signaling proteins, including TULA-2, a negative regulator of mast cell degranulation. J Biol Chem 287:8194–8204
Agrawal R, Carpino N, Tsygankov A (2008) TULA proteins regulate activity of the protein tyrosine kinase Syk. J Cell Biochem 104(3):953–964
Chen X, Ren L, Kim S, Carpino N, Daniel JL, Kunapuli SP, Tsygankov AY, Pei D (2010) Determination of the substrate specificity of protein-tyrosine phosphatase TULA-2 and identification of Syk as a TULA-2 substrate. J Biol Chem 285:31268–31276
Mikhailik A, Ford B, Keller J, Chen Y, Nassar N, Carpino N (2007) A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell 27:486–497
Smirnova EV, Collingwood TS, Bisbal C, Tsygankova OM, Bogush M, Meinkoth JL, Henderson EE, Annan RS, Tsygankov AY (2008) TULA proteins bind to ABCE-1, a host factor of HIV-1 assembly, and inhibit HIV-1 biogenesis in a UBA-dependent fashion. Virology 372:10–23
Collingwood TS, Smirnova EV, Bogush M, Carpino N, Annan RS, Tsygankov AY (2007) T-cell ubiquitin ligand affects cell death through a functional interaction with apoptosis-inducing factor, a key factor of caspase-independent apoptosis. J Biol Chem 282:30920–30928
Thomas DH, Getz TM, Newman TN, Dangelmaier CA, Carpino N, Kunapuli SP, Tsygankov AY, Daniel JL (2010) A novel histidine tyrosine phosphatase, TULA-2, associates with Syk and negatively regulates GPVI signaling in platelets. Blood 116:2570–2578
Brennan T, Adapala NS, Barbe MF, Yingling V, Sanjay A (2011) Abrogation of Cbl-PI3K interaction increases bone formation and osteoblast proliferation. Calcif Tissue Int 89:396–410
Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2:595–610
Miyazaki T, Takayanagi H, Isshiki M, Takahashi T, Okada M, Fukui Y, Oda H, Nakamura K, Hirai H, Kurokawa T, Tanaka S (2000) In vitro and in vivo suppression of osteoclast function by adenovirus vector-induced csk gene. J Bone Miner Res 15:41–51
Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM, Oda H, Nakamura K, Tanaka S (2000) Reciprocal role of ERK and NF-κB pathways in survival and activation of osteoclasts. J Cell Biol 148:333–342
Menaa C, Kurihara N, Roodman GD (2000) CFU-GM-derived cells form osteoclasts at a very high efficiency. Biochem Biophys Res Commun 267:943–946
Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R (2004) Src kinase activity is essential for osteoclast function. J Biol Chem 279:17660–17666
Sanjay A, Miyazaki T, Itzstein C, Purev E, Horne WC, Baron R (2006) Identification and functional characterization of an Src homology domain 3 domain-binding site on Cbl. FEBS J 273:5442–5456
Adapala NS, Barbe MF, Langdon WY, Nakamura MC, Tsygankov AY, Sanjay A (2010) The loss of Cbl-phosphatidylinositol 3-kinase interaction perturbs RANKL-mediated signaling, inhibiting bone resorption and promoting osteoclast survival. J Biol Chem 285:36745–36758
Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda T (1999) Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 190:1741–1754
Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL (2006) Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res (the official J Am Soc Bone Miner Res) 21:67–77
Mocsai A, Ruland J, Tybulewicz VL (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 10:387–402
Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP (2003) High dose M-CSF partially rescues the Dap12−/− osteoclast phenotype. J Cell Biochem 90:871–883
Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC (2004) The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA 101:6158–6163
Zou W, Kitaura H, Reeve J, Long F, Tybulewicz VL, Shattil SJ, Ginsberg MH, Ross FP, Teitelbaum SL (2007) Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol 176:877–888
Lupher ML, Rao N, Lill NL, Andoniou CE, Miyake S, Clark EA, Druker B, Band H (1998) Cbl-mediated negative regulation of the Syk tyrosine kinase. A critical role for Cbl phosphotyrosine-binding domain binding to Syk phosphotyrosine 323. J Biol Chem 273:35273–35281
Rao N, Ghosh AK, Ota S, Zhou P, Reddi AL, Hakezi K, Druker BK, Wu J, Band H (2001) The non-receptor tyrosine kinase Syk is a target of Cbl-mediated ubiquitylation upon B-cell receptor stimulation. EMBO J 20:7085–7095
Epple H, Cremasco V, Zhang K, Mao D, Longmore GD, Faccio R (2008) Phospholipase C gamma 2 modulates integrin signaling in the osteoclast by affecting the localization and activation of Src kinase. Mol Cell Biol 28:3610–3622
Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA (2002) Syk is required for integrin signaling in neutrophils. Immunity 16:547–558
Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T (2004) Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428:758–763
Lacey DL, Timms E, Tan H-L, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian Y-X, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang M-S, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan H-L, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Teitelbaum SL (2000) Bone resorption by osteoclasts. Science 289:1504–1508
Karsdal MA, Martin TJ, Bollerslev J, Christiansen C, Henriksen K (2007) Are nonresorbing osteoclasts sources of bone anabolic activity? J Bone Miner Res 22:487–494
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12:657–664
Kim HJ, Zhang K, Zhang L, Ross FP, Teitelbaum SL, Faccio R (2009) The Src family kinase, Lyn, suppresses osteoclastogenesis in vitro and in vivo. Proc Natl Acad Sci USA 106:2325–2330
Kim HJ, Warren JT, Kim SY, Chappel JC, DeSelm CJ, Ross FP, Zou W, Teitelbaum SL (2010) Fyn promotes proliferation, differentiation, survival and function of osteoclast lineage cells. J Cell Biochem 111:1107–1113
Acknowledgments
This work was supported by a grant from the Pennsylvania Department of Health to S. B and by the National Institute of Health Grant (AR055601) to A. S. The authors also thank Dr. Hector Aguila (Department of Immunology, University of Connecticut Health Center) for antibodies and help with FACS analysis. We also acknowledge the support of the MicroCT Core Facility at Temple University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2012_1203_MOESM1_ESM.pdf
Supplementary Figure 1. Osteoclast survival in in vitro assay is unaffected in the absence of TULA-2. Osteoclasts were derived from the bone marrow as described in the methods. Osteoclasts were either untreated or treated with RANKL (50 ng/ml) for 24 h. After treatment, cells were fixed and TRAP stained. The histogram represents the data expressed as percentage of initial number of TRAP + MNCs surviving after 24 h. (WT white bars; DKO black bars). * p < 0.05 as compared to untreated cells. (PDF 33 kb)
18_2012_1203_MOESM2_ESM.pdf
Supplementary Figure 2. DKO mice have decreased bone volume. Long bones from 12-week-old WT and DKO mice were X-rayed to qualitatively assess bone density. Insets in lower right of each panel shows the femur-tibia junction; arrows indicate sites with a qualitative decrease in bone density in the DKO samples. (PDF 30 kb)
18_2012_1203_MOESM3_ESM.pdf
Supplementary Figure 3. Controls for immunoprecipitation. Bone marrow macrophages (BMMs) were generated from the bone marrow of WT and DKO mice. BMMs were stimulated by cross-linking Fc receptor to activate the signaling pathway downstream of ITAM receptors. Lysates were used for immunoprecipitation, as described in Fig. 7. Controls used for immunoprecipitation are as follows: Lysis buffer with Anti-PTyr antibody (lane 1); cell lysates (50 μg) that were incubated with 1 μg of isotype control mouse IgG (lane 2) or that were either untreated (lane 3); control lysates (50 μg) from DKO osteoclasts (lane 4) or platelets (lane 5); and protein markers (lane 6). Blot was probed with phosphotyrosine antibodies. (PDF 204 kb)
Rights and permissions
About this article
Cite this article
Back, S.H., Adapala, N.S., Barbe, M.F. et al. TULA-2, a novel histidine phosphatase, regulates bone remodeling by modulating osteoclast function. Cell. Mol. Life Sci. 70, 1269–1284 (2013). https://doi.org/10.1007/s00018-012-1203-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1203-2