
Abstract
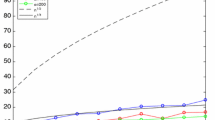
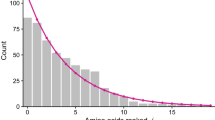
When two strings of symbols are aligned it is important to know whether the observed number of matches is better than that expected between two independent sequences with the same frequency of symbols. When strings are of different lengths, nulls need to be inserted in order to align the sequences. One approach is to use simple approximations of sampling for replacement. We describe an algorithm for exactly determining the frequencies of given numbers of matches, sampling without replacement. This does not lead to a simple closed form expression. However we show examples where sampling with, or without, replacement give very similar results and the simple approach may be adequate for all but the smallest cases.
Similar content being viewed by others
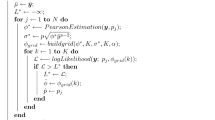
Literature
Altschul, S. F. and B. W. Erickson. 1988. Significance levels for biological sequence comparison using non-linear similarity functions.Bull. math. Biol. 50, 77–92.
Barker, W. C. and M. O. Dayhoff. 1977. Evolution of lipoproteins deduced from protein sequence data.Comp. Biochem. Physiol. 57B, 309–315.
Cedergren, R. J., D. Sankoff, B. LaRue and H. Grosjean. 1981. The evolving tRNA molecule.Crit. Rev. Biochem. 11, 35–104.
Felsenstein, J. 1982. Numerical methods for inferring evolutionary trees.Q. Rev. Biol. 57, 379–404.
Karlin, S., G. Ghandour and D. E. Foulser. 1985. DNA sequences comparisons of the human, mouse and rabbit immunoglobulin kappa gene.Molec. biol. Evol. 2, 35–52.
Kruskal, J. B. 1983. An overview of sequence comparison: time warps, string edits, and macromolecules.SIAM Rev. 25, 201–237.
Needleman, S. B. and C. D. Wunsch. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins.J. molec. Biol. 48, 443–453.
Peacock, D. 1982. Data handling for phylogenetic trees. InBiochemical Evolution, H. Gutfreund (ed.), pp. 85–115. Cambridge University Press.
Penny, D., L. R. Foulds and M. D. Hendy. 1982. Testing the theory of evolution by comparing phylogenetic trees constructed from five different protein sequences.Nature 297, 197–200.
Penny, D. and M. D. Hendy. 1985. Testing methods of evolutionary tree construction.Cladistics 1, 266–278.
Penny, D. and M. D. Hendy. 1986. Estimating the reliability of evolutionary trees.Molec. biol. Evol. 3, 403–417.
Waterman, M. S. 1984. General methods of sequence comparison.Bull. math. Biol. 46, 473–500.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Rinsma, I., Hendy, M. & Penny, D. Distribution of the number of matches between nucleotide sequences. Bltn Mathcal Biology 52, 349–358 (1990). https://doi.org/10.1007/BF02458576
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF02458576