
Abstract
The development of a new class of erythropoietin mimetic agents (EMA) for treating anemic conditions has been initiated with the discovery of oligopeptides capable of dimerizing the erythropoietin (EPO) receptor and thus stimulating erythropoiesis. The most promising amino acid sequences have been mounted on various different polymeric structures or carrier molecules to obtain highly active EPO-like drugs exhibiting beneficial and desirable pharmacokinetic profiles. Concomitant with creating new therapeutic options, erythropoietin mimetic peptide (EMP)-based drug candidates represent means to artificially enhance endurance performance and necessitate coverage by sports drug testing methods. Therefore, the aim of the present study was to develop a strategy for the comprehensive detection of EMPs in doping controls, which can be used complementary to existing protocols. Three model EMPs were used to provide proof-of-concept data. Following EPO receptor-facilitated purification of target analytes from human urine, the common presence of the cysteine-flanked core structure of EMPs was exploited to generate diagnostic peptides with the aid of a nonenzymatic cleavage procedure. Sensitive detection was accomplished by targeted-SIM/data-dependent MS2 analysis. Method characterization was conducted for the EMP-based drug peginesatide concerning specificity, linearity, precision, recovery, stability, ion suppression/enhancement, and limit of detection (LOD, 0.25 ng/mL). Additionally, first data for the identification of the erythropoietin mimetic peptides EMP1 and BB68 were generated, demonstrating the multi-analyte testing capability of the presented approach.

ᅟ
Similar content being viewed by others


Introduction
The elucidation of the cytokines pathway which, amongst others, can be assigned to interleukins, thrombopoietin, or erythropoietin (EPO), has facilitated the development of modern pharmaceuticals by emulating essential physiological factors [1, 2]. For a therapeutic enhancement of erythropoiesis, the spectrum of available drugs spans from low molecular HIF-stabilizers and obsolete cobalt administrations to high molecular mass drugs, including different forms of recombinant erythropoietin [3]. However, the distinct benefits in anemia treatment through erythroblast-increasing effects are subordinated in case of EPO misuse for endurance enhancement. Implementing the sophisticated Phage Display Technology (PDT) into pharmaceutical proceedings entails a fast and customized in vitro method to screen for highly specific active peptide regions that interact with physiological structures of interest [4]. The acquired peptide libraries establish the fund of the modern medicinal drug treasury and are utilized for e.g., full human antibody modeling and manufacturing [5, 6]. Fast evolving classes of bioactive substances pose unforeseen complications for analytical procedures and fast adaptable methods are desirable in sports drug testing. The common approach to enzymatically digest a congeneric group of proteins on a typical cleavage site with a subsequent determination of matching fragments (bottom-up determination) is susceptible to misinterpretation when these structures exhibit small and selective amino acid variations. This issue requires attention, particularly when small oligopeptides of analytical interest, e.g., erythropoietin mimetic peptides (EMP), are integrated in high molecular mass protein-based carriers or immunoglobulin structures. These designated EMPs are capable of mimicking the effect of EPO and exhibiting the desired EPO-receptor (EPOR) affinity after covalent dimerization of two peptidic monomers [7, 8]. The temporarily approved drug peginesatide (Omontys), the pre-release form of an EMP-carbohydrate product AGEM400(HES) [9], and the modified pipeline immunoglobulin CNTO530 [10] include slightly different EMP sequences modeled after the consensus sequence published by Wrighton et al. in 1996 [11]. Hitherto, two methods were implemented successfully into anti-doping analysis to reveal peginesatide abuses and based upon the use of bottom-up protein characterization by means of liquid chromatography/tandem mass spectrometry (LC-MS-MS) or immunoaffinity-purification and -detection [12, 13]. The presence of an inner cysteine-enclosed core in all currently pursued EMPs enables exploiting this feature for analytical purposes. In 1973, Jacobson et al. developed a cysteine-specific chemical cleavage of proteins at the amino acids alpha amide bond, which was optimized by Tang and Speicher in 2004 [14, 15]. The hydrolysis depends on the utilization of 2-nitro-5-thiocyanobenzoic acid (NTCB) under alkaline conditions with an additional high nucleophilic agent (glycine) generating a N-terminal acyliminothiazolidine (ait) residue. The previously mentioned erythropoiesis-stimulating drug candidates AGEM400(HES) and CNTO530 are representative for upcoming EMP structures, which can also be used as anemia therapeutics or misused for doping purposes to enhance endurance. Thereby, the lead compound of all erythropoietin mimetic peptides with the highest activity—EMP1—is integrated twice as a small part in CNTO530, a so called Mimetibody Technology product of Janssen Biotechnology (formerly known as Centocor) [16]. The EMPs of the active hydroxyethyl starch-polymer AGEM400(HES), the multi-bound EMP-dimer BB68, comprise an amino acid exchange similar to peginesatide (1-naphtyl alanine for tryptophane) [17]. With a growing EMP market, test methods largely independent from the drug’s support molecules are vital for modern doping controls. Hence, the EMPs’ capability to bind to the EPO-receptor was used for sample preparation and target analyte extraction prior to applying a low-cost chemical cleavage. Subsequent analysis was conducted with nano-liquid chromatography-electrospray ionization and high-resolution/high-accurate detection by utilizing quadrupole Orbitrap mass spectrometric equipment, which provided adequate sensitivity as demonstrated with the model substance peginesatide. Furthermore, first data concerning the detection of AGEM400(HES) and CNTO530 were generated by means of their EMP sequences, which served as surrogates in the absence of authentic drug reference material.
Experimental
Chemical and Reagents
The monomeric peptide of peginesatide, the Gly[15N, 13C2] labeled analog, as well as the BB68 and EMP1 monomers were obtained from BMFZ at the Heinrich-Heine University (Düsseldorf, Germany), and the active ingredient of the drug peginesatide—Omontys—was synthesized in-house [18]. The reagents for the cysteine-specific cleavage reaction in the form of 2-nitro-5-thiocyanobenzoic acid (NTCB), DL-dithiothreitol (DTT), glycine, sodium hydroxide, and the reagents for the synthesis COMU, Fmoc-iminodiacetic acid (F-IDAA), diisopropylethylamine (DIEA) and dimethylformamide (DMF) were purchased from Sigma Aldrich (Steinheim, Germany). The EPOR-Fc fusion protein, utilized in the prepurification steps was obtained from R&D Systems (Minneapolis, MN, USA). Additionally, the protein A/G beads with 10 mg/mL and the C18 spin columns for the desalting procedure were purchased from Pierce Protein Biology Products (Rockford, IL, USA). For the ultrafiltration processes, 15 mL 30 kDa MWCO centrifugal filter units from Merck-Millipore (Darmstadt, Germany) were utilized. Additionally, the ready-to-use tablets for the phosphate-buffered saline solution pH 7.4 (PBS), [100 mM sodium chloride, 80 mM Na2HPO4, 20 mM NaH2PO4 × 1 H2O] were also purchased from Sigma Aldrich (Steinheim, Germany). For all buffers and aqueous protein solutions deionized water of Milli-Q quality was utilized.
Synthesis of the Internal Standard (ISTD)
As reported in prior studies, a dimerization of EMP provides a significant increase towards the binding affinity at the EPOR [8]. Therefore, an appropriate internal standard for peginesatide with a similar receptor kinetic behavior was synthesized. In accordance with the dimeric pharmacophore of peginesatide, the labeled monomers were linked to COMU-activated Fmoc-iminodiacetic acid via the N-terminal lysine residue by generating an amide bond. To limit the extent of potential side reaction products, equal stoichiometric ratios of the monomer, COMU, and the linker with 2:2:1 were applied, and the synthesis was carried out in DMF. Herein, after a 1 h of pre-activating Fmoc-IDAA under alkaline DIEA conditions, the linker was added dropwise to the peptide. After 24 h reaction time, Fmoc was removed by adding an excess of piperidine. At the end of the synthesis, the dimer was purified by means of a semipreparative liquid chromatography apparatus equipped with an Eclipse XDB-C18 column (4.6 × 150 mm; Agilent Technologies, CA, USA). A detailed scheme with the LC-MS monitoring of the whole synthesis can be found in the Supplemental Information (Figure S-1).
Stock and Work Solutions
Working solutions containing peginesatide, the peginesatide- and BB68-monomer, and EMP1 were prepared from stock solutions (1 μg/mL) by dilution to concentrations of 1 ng/mL in deionized water. The stock solution of the internal standard, Gly[15N, 13C2]-labeled peginesatide monomer, also contained 1 μg/mL of the analyte, and was diluted 1:3 with deionized water to yield the corresponding working solution.
Sample Workup and EPOR Prepurification
For all specimens, 15 mL aliquots of fresh blank urine obtained from a healthy male volunteer were utilized. To enrich erythropoietin receptor agonists and diminish disturbing background interferences, an EPOR-based prepurification according to earlier published studies was carried out [19]. Prior to an ultrafiltration of urine and subsequent receptor-facilitated EMA enrichment, the urine was fortified with respective amounts of each analyte and transferred into the filter devices. After conducting the centrifugation and washing procedures, 3 μL of the ISTD working solution was added to the urine retentates. To adjust the final incubation volume to 1 mL per retentate, differences were compensated and accounted for with PBS. All receptor coated beads were treated with 100 μL of 5 mM BS3 in phosphate-buffered saline (pH 7.4) to avoid receptor co-elution. The prepared urine concentrates were incubated with the EPOR-Fc beads overnight at 4°C.
Cysteine-Specific Cleavage Procedure
After eluting the EMA enriched EPOR-beads twice with 50 μL of 2% acetic acid at 37°C for 30 min, the volume of the obtained slurry was reduced by means of a vacuum concentrator at 45°C. To a residue of approximately 10 μL, 2 μL of 20% ammonium bicarbonate solution was added to neutralize remnants of acetic acid. By adding 10 μL of 10 mM ammonium bicarbonate solution and 5 μL of fresh 0.01 M DTT, the disulfide bonds were reduced to their respective free thiols at 70°C for 30 min under constant stirring. Afterwards, the one pot cleavage procedure was completed via the addition of 20 μL of 1 M aqueous glycine solution, pH 9.5, and 16 μL of NTCB solution [1 mg/mL] directly into the specimen. The whole reaction was incubated for at least 16 h at 37°C. Owing to the mesomeric stabilization of the carboxy-nitrobenzenethiolate anion, the reaction progress could be monitored by an increase of the sample color from colorless to dark yellow (λ = 412 nm). A subsequent desalting procedure via C18 Spin Columns in accordance to the manufacturer’s protocol allowed for the removal of excess glycine, DTT, and NTCB prior to the LC-MS measurements. The purified extracts were evaporated and reconstituted in 50 μL of 2% acetic acid. The respective proposed molecular cleavage mechanism is shown in the Supplementary Information in Figure S-2.
LC-MS(/MS)
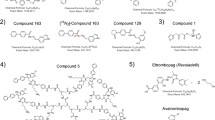
High resolution full scan MS measurements conducted during the ISTD synthesis with FWHM = 30.000 were accomplished using an Agilent Technologies (Waldbronn, Germany) HPLC coupled to an ABSciex (Darmstadt, Germany) TripleTOF 5600. The system was equipped with a Thermo Fisher Scientific (Dreieich, Germany) Accucore XL C8 (4 μm particle size, 100 × 3 mm) analytical column, and mobile phases consisted of aqueous formic acid 0.2%, pH 2 (solvent A) and acetonitrile as organic modifier (solvent B) for optimal ESI conditions. Thereby, the gradient decreased from 100% A to 10% A with 0.25 mL/min within 10 min. The subsequent re-equilibration time was 5 min. Method development for measuring the model compounds peginesatide, BB68, and EMP1 from human urine and respective method characterization studies were performed using a Waters (Eschborn, Germany) nanoAcquity liquid chromatography system coupled to a Thermo Fisher Scientific Q Exactive with positive nano-electrospray ionization. The system was operated with a PicoChip Reprosil-pur C18 nanospray column (New Objective, Woburn, MA, USA) with a particle size of 3 μm and dimensions of 100 mm × 0.75 μm. After the injection of 2 μL, a subsequent enrichment of the analytes was ensured via a trapping column over a period of 4 min by using a flow rate of 8 μL/min. As eluents, 0.1% (v/v) formic acid as aqueous (A) and acetonitrile/formic acid 0.1% as organic phase (B) were utilized. At a flow rate of 0.35 μL/min, gradient elution was chosen starting at 99% (A) decreasing to 30% (A) in 20 min. Followed by a 1-min wash phase with 10% (A), the column was re-equilibrated at starting conditions for 8 min. The MS setup consisted of a combination of selected ion monitoring (t-SIM) including the ions [M + 2H]2+ at m/z 568.75 for the analyte and 570.25 for the ISTD and data dependent MS2 (ddMS2) experiments. For the t-SIM experiments, the isolation window was set at 2 m/z. The cleavage products of the BB68 and EMP1 monomers yielded doubly protonated molecules at m/z 592.28 and 571.26. All mass-to-charge calculations based upon the exact mass of each analyte are shown in Figure 1. The resolution was set to 70.000 and 17.500 for the t-SIM selection and for the ddMS2, respectively. All ddMS2 experiments were acquired at a collision energy (CE) of 25 eV. To confirm the presence of the analytes, all determinations were performed in accordance to the WADA TD2010IDCR [20]. Thereby, the polyethylene glycol moieties of peginesatide were not part of the analysis.

Primary structures of (a) peginesatide, (b) isotopically labeled peginesatide, (c) EMP1 (CNTO530), and (d) BB68 [AGEM 400(HES)] according to the consensus sequence published by Wrighton et al. The enlarged sequence tags illustrate the location of the peptide cleavage sites and the resulting target analytes. Ac = Acetyl, Sar = Methylglycine, 1-nal = Naphtylalanine
Method Characterization
Method characterization was conducted for the model substance peginesatide regarding the parameters specificity, linearity, precision, recovery, stability, limit of detection (LOD), and ion suppression/enhancement in conformity with the WADA and FDA guidelines of analytical procedures [21, 22]. Showing the general applicability of the method for further analysis was one of the main proof-of-concept parameters.
Specificity
To probe for the method’s specificity, three ISTD-spiked blank urine concentrates of an initial volume of 15 mL underwent cysteine-specific cleavages and subsequent analysis as reported above. Additionally, another three aliquots spiked with 25 ng of peginesatide/mL were pretreated with aIGF1 beads to test for unspecific binding at magnetic beads [19]. Finally, three 15 mL aliquots containing peginesatide at 25 ng/mL were receptor-purified with EPOR-Fc beads and prepared for analysis.
Linearity
The linearity was tested using spiked urine samples by creating a calibration curve with samples spiked to 0.25, 1.0, 5.0, 10, and 25 ng/mL of peginesatide. Since no quantitative approach was intended, calibrators were analyzed only once.
Intraday Precision
Blank urine was fortified with 1, 10, and 25 ng peginesatide per mL and the obtained specimens were stored at 4°C until use. All concentration levels were prepared with 4 replicates of 15 mL each and analyzed via LC-MS to determine the intraday precision.
Recovery
Four aliquots of blank urine were spiked and worked up with 5 ng/mL peginesatide and 3 μL of ISTD in accordance to the EPOR prepurification protocol reported above. Simultaneously, another set of four samples of blank urine was treated the same way solely in the presence of 3 μL of ISTD without the addition of peginesatide. After the overnight incubation and washing steps, the same amount of 5 ng/mL peginesatide was spiked into the analyte-free retentates to resemble the samples of best-possible recovery (100%). Afterwards, all eight aliquots were subjected to the chemical cleavage procedure and the EPOR-specific extraction efficiency (recovery) was determined by comparing the signal responses of the target analytes. To probe for the completeness of the chemical cleavage, 1 ng/mL of pure peginesatide monomer was hydrolyzed. Afterwards, the monomer-to-acylisothiazolidine product ratio was determined by mass spectrometry, showing no remainders of the monomeric peginesatide sequences. Thus, an entire conversion of the peptides was assumed.
Stability
To exclude any influence of reagents or acidic conditions on the produced target analyte, in particular the acyliminothiazolidine residue, three aliquots of 10 μL peginesatide stock solution were hydrolyzed and measured directly and after 6 d. The samples were stored airtight at 10°C in Eppendorf Cap tubes until re-analysis.
LOD
Three replicates at 0.1 ng/mL, 0.25 ng/mL, and 0.5 ng/mL each were utilized to determine the limit of detection. Thereby, the signal to noise ratio (S/N) of >3 had to be fulfilled.
Ion Suppression/Enhancement
To test for a potential influence of the matrix or the remaining chemicals on the peak area intensity, three blank urine EPOR-purified eluates, 1 M glycine/NTCB solution aliquots, and three samples of pure water were fortified with 0.45 pmol/mL of cleaved and C18 purified peginesatide reference (equally 10 ng/mL of intact peginesatide). The subsequent measurements were assessed concerning increasing or decreasing signal responses.
Results
Owing to the partial elimination of peginesatide into urine after parenteral application, doping control urine samples represent a viable matrix for sports drug testing concerning this EMA [23]. In consideration of intended therapeutic dosages of 0.04 mg/kg body weight and reported cumulated urinary concentrations of approximately 70% of the initial single dose after 336 h, the herein used spiked amounts of the analyte simulate authentic levels [24, 25].
LC-MS/MS
As shown in Figure 1, the acylisothiazolidine products of the model substances comprise substantial sequence homologies, all of which are unequivocally differentiated by the conducted ddMS2 experiments yielding characteristic dissociation patterns. The expected and observed ion transitions for peginesatide and the corresponding ISTD are listed in detail in Table 1. The doubly charged molecules at m/z 568.7539 and 570.2550 (±2.5 ppm), respectively, were used as precursor ions producing a series of diagnostic a, b, and y′′ product ions that conclusively corroborated the assigned amino acid sequences of the analytes (Figure 2). In addition, peginesatide and BB68 comprise the artificial amino acid 1-naphthylalanine, a modification commonly used to lower enzymatic degradation of therapeutic peptides in vivo [23], giving rise to the product ion at m/z = 170.0964. For semiquantitative analytical purposes, the precursor ion plus additional information as obtained from the two most significant ion transitions (568.754-170.096; 568.754-747.306) was utilized.

Product ion spectra of the 2-fold protonated precursor of (a) peginesatide and the glycine-[15N, 13C2]-labeled peginesatide (m/z 568.75 and 570.25), (b) EMP1 (m/z 571.26), and (c) the BB68 monomer (m/z 592.28) with respective extracted ion chromatograms. Further, a structure for product ion at m/z 170.0964 is suggested as a result of the non-natural amino acid 1-nal indicated by an asterisk in the spectra
Analytical Method Characterization
Method characterization for doping control purposes was conducted exclusively with the model substance peginesatide yielding the data summarized in Table 2. Reason for excluding EMP1 and BB68 into the assay characterization was the nonavailability of the required dimeric forms and the fact that therapeutic formulations of CNTO530 and AGEM400(HES) were not available for comprehensive studies. Nevertheless, the respective monomeric EMPs, which represent the active principles and target analytes, were subjected to the cysteine-specific cleavage and subsequent analysis confirming the expected conversion into compounds unequivocally identifiable in doping control samples.
Specificity
The specificity of the method was demonstrated by the absence of interfering signals in blank urine specimens (Figure 3). Further, no binding occurred at aIGF1 beads and no signal was observed for blank urine samples when applying the established EPOR-Fc beads-based extraction. In contrast, peginesatide-spiked urine aliquots yielded the expected signals and precursor/product ion pairs attributed to the peptide sequence resulting from the cysteine-specific hydrolysis.

Targeted selected ion monitoring (t-SIM) chromatograms for an ISTD-fortified blank urine (left) and a urine sample containing 0.25 ng/mL of peginesatide (right)
Linearity
Prior to characterizing the performance of the entire method, the independence of the chemical hydrolysis reaction from analyte concentration levels was assessed. Here, optimized conditions concerning duration, temperature, and stoichiometry were used by omitting the ultrafiltration and EPOR purification step, and the response of the analyte signal showed an acceptable linear approximation (data not shown). Also in urinary matrix, a linear correlation between analyte concentration and signal abundance was determined in the defined working range of 0.25–25 ng/mL. The coefficient of correlation was 0.986 with a slope of 0.0249 and an intercept with 0.0115. Above 50 ng/mL, the assay indicated saturation of the binding capacity of the employed 2.5 μg (approximately 0.042 nmol) of the EPOR-Fc per specimen (see Supplementary Information, Figure S-4). Therefore, at higher concentrations, nonlinear characteristics were found to dominate the concentration–response correlation.
Precision
For all concentration levels at 1, 10, and 25 ng/mL, the means and relative standard deviations (RSD) corrected to the ISTD signal response were calculated. A 4-fold determination of each spiked urine aliquot resulted in RSD values of 18.3%, 9.7%, and 13.3%, respectively.
Recovery
The analysis of the target peptide obtained from peginesatide monomer revealed negligible amounts of intact monomeric substance. Subjecting the PEGylated homodimeric drug to chemical hydrolysis, similar signal responses were observed, suggesting a cleavage yield of ≥90% after 16 h. The overall mean recovery including ultrafiltration, EPOR-based extraction, and hydrolysis was determined with approximately 40% (Table 2). Thereby, the relative standard deviation was 17.3%.
Stability
After 6 days, the mean peak areas of the target analyte measured in the three specimens decreased to 89% of the initial abundance, indicating a sufficient stability of the analyte (and the generated acylisothiazolidine residue) in vials under benchtop conditions.
Limit of Detection (LOD) and Ion Suppression/Enhancement Effects
The limit of detection was set at 0.25 ng/mL after the unambiguous determination of the acylisothiazolidine monomer signal at this concentration level (Figure 3). The signal to noise ratios of at least two precursor–product ion pairs exceeded the required value of 3 [20, 26]. In addition, no significant matrix effects resulting in ion suppression or enhancement were observed.
Discussion
To date, chemical approaches for hydrolyzing peptides and proteins with high specificity at defined amino acid peptides, in particular for subsequent bottom-up analyses of compounds in biological matrices, are rarely used [27, 28]. This is arguably due to a great variety of available enzymes for peptide/protein analytics, which proved robust and easy-to-handle by employing mostly physiological conditions. In contrast, the chemical hydrolysis of peptidic compounds commonly requires harsh conditions that can cause unpredictable and unspecific reaction by-products. In the present study, conditions of common enzymatic digestion protocols are reflected by using reducing agents such as DTT at 70°C and hydrolysis temperatures of 37°C. The obtained N-terminally acylisothiazolidine-derivatized peptides as derived from the cysteine-specific cleavage facilitate the detection of target analytes because there is no concordance with any known naturally occurring amino acid sequence in humans. Therefore, coincidentally enzyme-originated or renally excreted peptides with similar masses and amino acid sequences are excluded, in particular when considering characteristic a, b, and y′′ product ions generated in MS2 experiments. In comparison to existing methods for EMP detection in sports drug testing, the herein presented approach is considerably more time-consuming but allows for expanded comprehensiveness for this class of prohibited substances. In addition, supporting evidence for the presence of physiologically relevant peptides detected in doping control specimens is provided by the preceding EPOR-based enrichment and purification of analytes. To date, the assays’ area of application does not include quantitative analysis. The quantitative analysis of peginesatide has been accomplished earlier using alternative approaches [13, 18]; however, these methods are dedicated to one erythropoietin mimetic only and do not allow the immediate inclusion of additional related analytes. Moreover, cysteines represent moieties of less frequent occurrence and, thus, the chemical proteolysis can support generating larger peptide segments via cysteine-specific cleavage. Enzymes, as natural catalysts, have the ability to accelerate substrate conversion by simultaneously keeping their proteolytic activity after the reaction. In case of chemical cleavage, reactants are consumed, necessitating an excess in each sample that potentially affects the performance of the LC-MS analysis. Hence, the isolation and prepurification of the target analytes prior to the bottom-up analytics are indispensable, and desalting is recommended. A disadvantage of this study is the nonavailability of elimination study urine specimens collected after peginesatide administration as a reason of the final recall of peginesatide in 2013. Nevertheless, the disposability of black market aliquots of Omontys can be assumed.
Conclusion
The herein presented approach demonstrates the efficiency of chemical cleavage and its utility in doping controls, complementing an enzymatic hydrolysis-based test. Erythropoietin mimetic peptides can be seen as a pharmaceutical benchmark for PDT-created peptidic drugs and drug candidates presenting with high therapeutic potency while exhibiting nonconforming properties compared with its physiological counterpart EPO. Pinpointing and exploiting conserved structural features of the target analytes are the basis of the utilized analytical method. Therefore, the cysteine-entrapped core structure of EMPs provides an excellent motif. Moreover, the carrier/drug support structure of the embedded oligopeptide is irrelevant as the cysteine cleavage delivers sufficient information for LC-MS/MS determinations to detect segments indicating and evidencing the presence of EMP-derived molecules. The expected therapeutic dosages, serum concentrations, and physiologically excreted amounts of modern EMP-derived drugs suggest that the herein presented LOD with 0.25 ng/mL will serve doping control analytical purposes.
References
O'Shea, J.J., Gadina, M., Kanno, Y.: Cytokine signaling: birth of a pathway. J. Immunol. 187, 5475–5478 (2011)
Tsiftsoglou, A.S., Vizirianakis, I.S., Strouboulis, J.: Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 61, 800–830 (2009)
Macdougall, I.C.: Novel erythropoiesis-stimulating agents: a new era in anemia management. Clin. J. Am. Soc. Nephrol. 3, 200–207 (2008)
Smith, G.P.: Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 (1985)
Deantonio, C., Cotella, D., Macor, P., Santoro, C., Sblattero, D.: In: Steinitz. M. (ed.) Human Monoclonal Antibodies, vol. 1060, pp. 277–295. Humana Press, New York City (2014)
Nelson, A.L., Dhimolea, E., Reichert, J.M.: Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 (2010)
Livnah, O., Stura, E.A., Johnson, D.L., Middleton, S.A., Mulcahy, L.S., Wrighton, N.C., Dower, W.J., Jolliffe, L.K., Wilson, I.A.: Functional mimicry of a protein hormone by a peptide agonist: the EPO receptor complex at 2.8 A. Science 273, 464–471 (1996)
Wrighton, N.C., Balasubramanian, P., Barbone, F.P., Kashyap, A.K., Farrell, F.X., Jolliffe, L.K., Barrett, R.W., Dower, W.J.: Increased potency of an erythropoietin peptide mimetic through covalent dimerization. Nat. Biotechnol. 15, 1261–1265 (1997)
Kessler, C., Greindl, A., Breuer, B., Haberl, U., Rybka, A., Emgenbroich, M., Frank, H.G., Potgens, A.J.: Erythropoietin mimetic compound AGEM400(HES) binds to the same receptor as erythropoietin but displays a different spectrum of activities. Cytokine 57, 226–237 (2012)
Bugelski, P.J., Capocasale, R.J., Makropoulos, D., Marshall, D., Fisher, P.W., Lu, J., Achuthanandam, R., Spinka-Doms, T., Kwok, D., Graden, D., Volk, A., Nesspor, T., James, I.E., Huang, C.: CNTO 530: molecular pharmacology in human UT-7EPO cells and pharmacokinetics and pharmacodynamics in mice. J. Biotechnol. 134, 171–180 (2008)
Wrighton, N.C., Farrell, F.X., Chang, R., Kashyap, A.K., Barbone, F.P., Mulcahy, L.S., Johnson, D.L., Barrett, R.W., Jolliffe, L.K., Dower, W.J.: Small peptides as potent mimetics of the protein hormone erythropoietin. Science 273, 458–464 (1996)
Moller, I., Thomas, A., Delahaut, P., Geyer, H., Schanzer, W., Thevis, M.: Mass spectrometric detection of peginesatide in human urine in doping control analysis. J. Pharm. Biomed. Anal. 70, 512–517 (2012)
Leuenberger, N., Saugy, J., Mortensen, R.B., Schatz, P.J., Giraud, S., Saugy, M.: Methods for detection and confirmation of hematide/peginesatide in anti-doping samples. Forensic Sci. Int. 213, 15–19 (2011)
Jacobson, G.R., Schaffer, M.H., Stark, G.R., Vanaman, T.C.: Specific chemical cleavage in high yield at the amino peptide bonds of cysteine and cystine residues. J. Biol. Chem. 248, 6583–6591 (1973)
Tang, H.-Y., Speicher, D.W.: Identification of alternative products and optimization of 2-nitro-5-thiocyanatobenzoic acid cyanylation and cleavage at cysteine residues. Anal. Biochem. 334, 48–61 (2004)
Picha, K., Huang, C., Bugelski, P., O’Neil, K.: In: Nixon, A.E. (ed.) Therapeutic Peptides, vol. 1088, pp. 125–145. Humana Press, New York City (2014)
Greindl, A., Kessler, C., Breuer, B., Haberl, U., Rybka, A., Emgenbroich, M., Pötgens, A.J.G., Frank, H.-G.: AGEM400(HES), a novel erythropoietin mimetic peptide conjugated to hydroxyethyl starch with excellent in vitro efficacy. Open Hematol. J. 4, 1–14 (2010)
Moller, I., Thomas, A., Geyer, H., Schanzer, W., Thevis, M.: Synthesis, characterisation, and mass spectrometric detection of a pegylated EPO-mimetic peptide for sports drug testing purposes. Rapid Commun. Mass Spectrom. 25, 2115–2123 (2011)
Vogel, M., Blobel, M., Thomas, A., Walpurgis, K., Schanzer, W., Reichel, C., Thevis, M.: Isolation, enrichment, and analysis of erythropoietins in anti-doping analysis by receptor-coated magnetic beads and liquid chromatography-mass spectrometry. Anal. Chem. 86, 12014–12021 (2014)
WADA, Identification Criteria for Qualitive Assays in Column Chromatography and Mass Spectrometry. available at: https://wada-main-prod.s3.amazonaws.com/resources/files/WADA_TD2010IDCRv1.0_Identification%20Criteria%20for%20Qualitative%20Assays_May%2008%202010_EN.doc.pdf. Accessed 27 May 2015 (2010)
Administration, F. a. D. Bioanalytical Method Validation. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf. Accessed 27 May 2015 (2013)
WADA, International Standard for Laboratories. Available at: https://wada-main-prod.s3.amazonaws.com/resources/files/WADA_Int_Standard_Laboratories_2012_EN.pdf. Accessed 27 May 2015 (2012)
Stead, R.B., Lambert, J., Wessels, D., Iwashita, J.S., Leuther, K.K., Woodburn, K.W., Schatz, P.J., Okamoto, D.M., Naso, R., Duliege, A.M.: Evaluation of the safety and pharmacodynamics of Hematide, a novel erythropoietic agent, in a phase 1, double-blind, placebo-controlled, dose-escalation study in healthy volunteers. Blood 108, 1830–1834 (2006)
Affymax Incorporation. Omontys–Highlights of Prescribing Information. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202799s000lbl.pdf. Accessed 27 May 2015 (2012)
Woodburn, K.W., Fong, K.L., Wilson, S.D., Sloneker, S., Strzemienski, P., Solon, E., Moriya, Y., Tagawa, Y.: Peginesatide clearance, distribution, metabolism, and excretion in monkeys following intravenous administration. Drug Metab. Dispos. 41, 774–784 (2013)
WADA, Minimum Required Performance Levels for Detection and Identification of Non-Threshold Substances. Available at: https://wada-main-prod.s3.amazonaws.com/resources/files/WADA-TD2014MRPL-v1-Minimum-Required-Performance-Levels-EN.pdf. Accessed 27 May 2015 (2014)
Crimmins, D.L., Mische, S.M., Denslow, N.D.: Current Protocols in Protein Science, 11.4.1-11.4.11. John Wiley and Sons, Inc., Hoboken (2001)
Smith, B.: In: Walker, J. (ed.) Basic Protein and Peptide Protocols, vol. 32, pp. 297–309. Humana Press, New York City (1994)
Acknowledgments
This study was carried out with support of the World Anti-Doping Agency (grant #14B03MT), Montreal, Canada, the Manfred Donike Institute for Doping Analysis, Cologne, Germany, and the Federal Ministry of the Interior of the Federal Republic of Germany, Berlin, Germany.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Information
(DOCX 409 kb)
Rights and permissions
About this article

Cite this article
Vogel, M., Thomas, A., Schänzer, W. et al. EPOR-Based Purification and Analysis of Erythropoietin Mimetic Peptides from Human Urine by Cys-Specific Cleavage and LC/MS/MS. J. Am. Soc. Mass Spectrom. 26, 1617–1625 (2015). https://doi.org/10.1007/s13361-015-1189-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1189-8