
Abstract
Although complete remissions can be achieved in most patients younger than 60 years of age with untreated acute myeloid leukemia (AML), only 30–40 % of patients remain long-term survivors. Furthermore, long-term survivors represent only 10–15 % of all AML patients older than 60 years of age and <10 % of all patients with relapsed AML. The development of new treatments for AML is therefore needed. Novel therapies should target specific mechanisms and pathways implicated in the development and maintenance of AML, should strive to have better tolerability than conventional combination chemotherapy, be associated with improved quality of life and minimize utilization of health care resources. In this manuscript, we discuss the role of epigenetic regulators and immunomodulatory agents in the treatment of AML. Also, we review the data on inhibitors of protein homeostasis and its synergistic effect to DNA methyltransferase inhibitors, the potential role for inhibitors of heat shock proteins and the mitotic machinery and a novel formulation of conventional chemotherapeutic agents given at a fixed molar concentration. Finally, we briefly share our views on optimal clinical trial design and patient selection for future studies in AML.
Similar content being viewed by others

Background
It is estimated that nearly 14000 adults will be diagnosed with acute myeloid leukemia (AML) in the US in 2012, and about 10000 will die from leukemia (SEER database, NCI). The fact that the median age at diagnosis ranges between 67 and 70 years, significantly complicates therapeutic options because of the reduced ability of elderly patients to tolerate intensive combination chemotherapy and the high incidence of co-morbid conditions. Despite advances in supportive care and optimization of treatment regimen, the 5-year survival for adults with AML is only about 20–25 % (SEER database NCI). The treatment of patients over the age of 60 is particularly challenging and results in median survival of <1 year with low rates of complete responses (CR) to therapy and high relapse rates in those who do achieve a CR. Adult patients with AML who are candidates for intensive therapy are treated with the combination of cytarabine and daunorubicin (or idarubicin), a regimen that was first introduced in the 1970s and is typically followed by three to four cycles of consolidation with high dose cytarabine or allogeneic hematopoietic stem cell transplantation. Recent studies have shown improved outcomes with intensified doses of daunorubicin, highlighting that ongoing optimization of standard treatments can lead to substantial improvements in the treatment of acute leukemias [1]. Patients who are not expected to tolerate intensive chemotherapy, such as individuals over the age of 60, often receive less intensive regimen or investigational agents [2].
It should be noted that an important exception to the above-mentioned statistics is acute promyelocytic leukemia (APL). This type of AML, formerly designated as M3, has a much better prognosis due to the implementation of sensitive molecular diagnostic tools and introduction of all-trans retinoic acid and arsenic trioxide into clinical practice. APL will not be covered in this review and the reader is referred elsewhere for review of this literature [3, 4].
Patients with AML can be stratified based on characteristic cytogenetic abnormalities into groups with poor, intermediate, and good prognosis. This classification has been further refined by the testing for specific molecular abnormalities in the NPM1, FLT3 and CEBPΑ genes [2]. Recent high-throughput sequencing efforts have identified recurrent mutations in DNMT3A, TET2, IDH1/2 among other genes that may allow further prognostic and therapeutic stratification of AML patients [2, 5–8]. Improved prognostic stratification based on molecular abnormalities and a detailed understanding of the mechanisms of leukemogenesis may allow hematologists to more precisely tailor available therapy to individual patients and lead to the identification of new therapeutic targets. One long-term goal of such research is to develop therapies that selectively eliminate AML cells and thereby cause less harmful side effects to the patient. Further genomic characterization of AML may also provide crucial insight into the molecular differences between younger and older patients with AML that has already been described at a cytogenetic level [9, 10].
The tremendous advances in the understanding of the molecular biology and biochemistry of hematopoiesis and transformation to leukemia have lead to the identification of numerous putative therapeutic targets and a growing number of promising investigational agents. In this review, we describe some of the novel agents currently being used and tested in the treatment of AML that we find particularly promising (Fig. 1). Selected active clinical trials exploring these novel agents are listed on Tables 1 and 2.

Schematic representation of the mechanism of action for novel agents for the treatment of AML. HDAC histone deacetylase, DNMT DNA methyltransferase, NAE Nedd8 activating enzyme
Lenalidomide
The therapeutic efficacy of the thalidomide derivative, lenalidomide, was first demonstrated in multiple myeloma patients and has lead to its investigation in other hematolymphoid malignancies [11]. The mechanism of action of lenalidomide is not clearly established but it is thought to include direct anti-tumor effects, changes in the bone marrow microenvironment, inhibition of angiogenesis, and immunomodulation [12]. High dose single agent lenalidomide treatment of 50 mg daily for up to two 28-day cycles in 33 patients over the age of 60 with previously untreated AML resulted in complete response/complete response with incomplete count recovery (CR/CRi) in 30 % of patients. Patients with stable disease or improvement were then given up to 12 months of lenalidomide 10 mg daily until disease progression or development of intolerable side effects. The most common serious adverse events included bone marrow suppression and infection [13]. The role of newer immunomodulating drugs such as pomalidomide in the treatment of AML has not been established but merits further investigation.
Epigenetic modulators
Epigenetic gene regulation refers to the control of transcription that is not inherited via the DNA sequence but rather by other means such as DNA methylation or a variety of histone modifications including acetylation, methylation and phosphorylation (Fig. 2) [14]. Abnormalities in the epigenetic landscape of tumor cells are increasingly being appreciated and are highlighted by the frequency of mutations in enzymes encoding methyltransferases and other epigenetic regulators in cancers including AML, where DNMT3A was found to be recurrently mutated by high-throughput DNA sequencing [5, 14, 15]. Furthermore, epigenetic changes are thought to be one of the key mechanisms of treatment resistance in human malignancies [16].

Target for epigenetic regulators in the treatment of AML. HDAC histone deacetylase, DNMT DNA methyltransferase, αKG alpha-ketoglutarate
Promoter hypermethylation is one putative mechanism thought to lead to the silencing of expression of key tumor suppressors in human malignancies, and reversal of this epigenetic modification is one of the proposed mechanisms of action of the FDA approved DNA methyltransferase (DNMT) inhibitors azacitidine and decitabine. Both drugs have been used in the treatment of older AML patients with azacitidine 75 mg/m2 for 7 days per 28-day cycle resulting in a CR rate of 20 %, and 10 days of decitabine 20 mg/m2 treatment leading to a CR in 47 % of patients treated with a median of three cycles. The main side effect of azacitidine and decitabine treatment is bone marrow suppression and the sequelae associated with anemia, thrombocytopenia, and neutropenia [17–20].
An attractive theoretical application of DNMT inhibitors is to prime leukemia cells to induce expression of epigenetically silenced tumor suppressor genes, and thereby sensitize the cancer cells to traditional antineoplastic agents. This approach was tested in a phase I trial where 3, 5, or 7 days of decitabine 20 mg/m2 were given prior to standard AML induction chemotherapy with daunorubicin and cytarabine. Toxicity was generally similar to that of induction chemotherapy alone and a 5-day priming regimen was chosen for subsequent phase II studies [21].
Standard dosed azacitidine has also been combined safely with sequential lenalidomide 50 mg PO daily for 3 weeks followed by 2 weeks off therapy in 6-week cycles. The combined therapy was generally well tolerated and resulted in a CR/CRi rate of 44 % in elderly AML patients and is now under investigation in further trials [22]. The development of an oral azacitidine formulation raises the possibility of an outpatient oral regimen for patients with AML who are not candidates for intensive chemotherapy [23]. Oral azacitidine was tested in myelodysplastic syndrome, chronic myelomonocytic leukemia as well as AML and based on promoter methylation status, oral administration was found to have slightly attenuated but similar biological activity when compared to parenteral azacitidine. If this difference is found to be therapeutically relevant in patients, it may be overcome by different dosing regimen of oral azacitidine [24]. Future trials investigating the role of oral azacitidine with other oral agents in a larger cohort of patients are planned for the near future.
Acetylation of histone proteins can promote transcription of specific genes, and histone deacetylases have been shown to be involved in concert with methyltransferases in silencing tumor suppressor genes. Romidepsin and vorinostat are two HDAC inhibitors (HDACi) that are approved for the treatment of peripheral (romidepsin) and cutaneous (romidepsin and vorinostat) T-cell lymphomas [25, 26]. The mechanism of action of HDACi in AML and other human malignancies is incompletely understood. The challenge in determining the precise molecular effects of HDACi is complicated in part by the presence of eleven different zinc-dependent HDAC genes in humans setting the stage for enzymatic redundancy and specialized functions of specific isoforms [27]. Several novel agents targeting HDACs are currently in development (such as panobinostat, belinostat, MS-275) for the treatment of various malignancies. Furthermore, combinations of HDACi with DNMT inhibitors have shown synergistic effects in reversing histone deacetylation [28]. This has provided a scientific rationale for ongoing clinical trials testing the safety and efficacy of combined HDACi (vorinostat, belinostat, panobinostat) and azacitidine/decitabine in AML with some trials adding gemtuzumab ozogamicin as well [29]. The addition of vorinostat to standard induction therapy with cytarabine and idarubicin was recently evaluated in a phase II trial that included 68 patients with AML. The investigators found no vorinostat-related toxicity and therefore will pursue a randomized trial investigating the efficacy of adding vorinostat to standard induction versus standard therapy alone [30].
Nedd8 activating enzyme (NAE) inhibitors
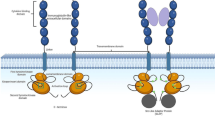
Ubiquitin and ubiquitin-like protein (UBL) pathways are key secondary modifications regulating protein stability and function (Fig. 3). The complexity of these pathways as well as their role in cancer cell growth and survival is increasingly being appreciated, and targeting of UBL in human malignancies has become an intense area of investigation. All but one cullin-RING E3 ubiquitin ligases are dependent on neddylation by the NEDD8-activating enzyme for full activation. These enzymes catalyze the last step in ubiquitylation, thereby imparting substrate specificity and regulating diverse cellular processes such as signal transduction and cell cycle dynamics [31]. The Nedd8-activating enzyme inhibitor MLN4924 has been tested in relapsed or refractory AML based on promising preclinical studies [32]. A phase I clinical trial is ongoing with preliminary results indicating single agent activity in AML that was presented in abstract form (Swords et al. ASH, 2010). Grade 3 or higher toxicities of MLN4924 included neutropenia, liver function abnormalities, and pneumonia. MLN4924 prevents cullin-RING E3 ligases by forming an adduct with NEDD8, a complex that was recently experimentally demonstrated in primary bone marrow samples from patients with AML treated with MLN4924 using immunohistochemical stains (MacDonald et al., EHA 2012). Pharmacologic modulation of the neddylation–ubiquitylation pathways in AML is of particular interest since this pathway is not targeted by other medications currently used in the treatment of AML and may therefore provide synergistic anti-leukemic activity.

Proposed mechanism of action for the Nedd8 activating enzyme inhibitor. NAE Nedd8 activating enzyme, Ubc12 E2 conjugating enzyme for neddylation pathway, Ub ubiquitin
Hsp90 inhibitors
Hsp90 is one member of a group of proteins known as heat shock proteins, a classification that is based on their increased expression in response to cellular stress such as heat, heavy metal exposure, acidosis, and hypoxia. Most heat shock proteins are expressed at significant levels in normal physiological conditions and serve a multitude of functions in cellular biology including protein folding, translocation of proteins across membranes, quality control in the endoplasmic reticulum, and normal protein turnover. Interestingly, these protein chaperones also play a role in more complex processes such as post-translational modification of signaling molecules and the assembly of large protein complexes as well as processing of peptides by the immune system for antigen presentation [33]. Hsp90 is not required for the synthesis of most polypeptides but instead stabilizes cellular proteins that are conformationally labile and control cell growth, survival, and tissue development. Hsp90 is known to interact with a large number of client proteins, which include receptor tyrosine kinases, telomerase, and Akt among others. The observation that Hsp90 and other molecular chaperones are overexpressed in human malignancies has lead to the hypothesis that they stabilize oncogenically mutated, labile proteins in cancer cells and generally protect those cells from a hostile environment. Hsp90 function is dependent on ATP hydrolysis, which makes it vulnerable to targeting with small molecules that bind the ATP-binding pocket [33]. A concern for targeting HSP90 is the possibility of serious adverse effects due to the ubiquitous nature of this chaperone’s function; however, the use of proteasome inhibitors has been highly effective in multiple myeloma and highlights that targeting of key cellular pathways involved in protein degradation is indeed possible in patients without excessive toxicity.
The high incidence of activation of Hsp90 client proteins such as Flt3, c-Kit, Akt, and mitogen-activated protein kinase in AML has provided a scientific rationale for early phase trials of alvespimycin and ganetespib [34]. A phase I study of alvespimycin in 24 patients with a median prior treatment of two induction regimen established a dose of 24 mg/m2 IV twice weekly for phase II studies and showed promising single agent activity with 3 patients having achieved a CRi. Two of these patients had chromosome 7q deletions while the other had normal cytogenetics. Flt3 mutation status was unknown for one of these patients and negative for the other two patients who had a CRi. The responses were seen after one cycle of alvespimycin and were maintained for 2, 5, and 11 cycles in the three patients. Dose limiting toxicity was reached at 32 mg/m2 where cardiac toxicity developed in two patients. Common side effects included neutropenia, fever, nausea and diarrhea [34]. A phase I/II trial of ganetespib has also been conducted but results have not yet been reported (http://www.clinicaltrials.org).
Cpx 351
CPX-351 is a liposomal-based experimental drug that results in the delivery of a 5:1 ratio of cytarabine to anthracycline that is maintained for up to 24 h in the bone marrow and blood of leukemia bearing mice and has been shown to result in optimal synergy between these two drugs in preclinical models [35]. The initial experience of CPX-351 in the treatment of 48 patients with relapsed or refractory MDS/AML defined the MTD at 101 U/m2 when given on days 1, 3, and 5 of an induction cycle, and nine out of the 43 patients with AML achieved a CR. Dose-limiting toxicities included hypertensive crisis, congestive heart failure, and prolonged cytopenias [36]. Phase II trials investigating the use of CPX-351 versus standard cytarabine/daunorubicin in elderly patients and CPX-351 versus intensive salvage therapy in relapsed disease in patients under the age of 60 have recently been completed. In previously untreated patients, CPX-351 treatment was associated with higher CR rates, event-free and overall survival and lower rate of 30- and 60-day mortality (Lancet, ASH, 2010). These results were particularly encouraging in elderly patients with secondary AML. Similar results have been observed in patients receiving CPX-351 as first salvage, with particular survival benefit to those patients with unfavorable European Prognostic Index (EPI) risk. (Goldberg et al. ASCO, 2012).
Aurora B kinase inhibitor
The human Aurora kinase family includes three members designated A, B, and C, of which Aurora A kinase has the strongest association with carcinogenesis in humans. The Aurora B kinase is a component of the chromosomal passenger complex and thereby plays a crucial regulatory role in chromosomal dynamics during mitosis, suggesting that it may also be a suitable cancer therapeutic target [37]. Preclinical studies with the Aurora B selective small molecule inhibitor AZD1152 in murine xenotransplantation experiments involving AML cell lines and primary AML cells showed inhibitory effects on the leukemia cell growth [38]. Based on the promising activity seen in the preclinical studies, a phase I/II trial of AZD1152 infused continuously for 7 days out of a 21-day cycle in the treatment of newly diagnosed or relapsed AML was initiated. A MTD of 1200 mg was established and resulted in a CR/CRi of 18 % in a cohort of 32 patients with AML. Among the more common serious side effects were febrile neutropenia and mucositis [39]. A subsequent study combining low dose cytarabine with AZD1152 has been completed but results have not yet been published.
Improving clinical trial design in AML
The historic response (1991–2002, NCI) rate for first-in-human clinical trials is about 5 % while phase I trials in general resulted in a response rate of about 10 % [40]. Current late-phase clinical trials are based on testing novel combination of existing drugs or the addition of new drugs to standard therapy in large cohorts of patients to establish efficacy. These large clinical trials can take significant time to accrue the necessary number of patients, thereby delaying the approval of new therapies. The genetic diversity of AML is increasingly being appreciated in the era of large-scale genetic analyses that has allowed the genomic characterization of AML at a previously unprecedented rate and depth. While it is unlikely that any one novel agent, particularly more targeted drugs, will work in the vast majority of cases of adult AML, many agents are likely to be active in a subset of patients. The integration of genomic analysis into future clinical trials may help to identify the subset of patients who will respond to specific agents or combinations of medications and thus allow for smaller, more efficient clinical trials. Furthermore, those exact studies may also be able to pinpoint mechanisms of resistance to specific treatments, identify key molecular targets that predict response to certain drugs, and may be targets for future therapies [41]. This approach will depend on collaborative efforts between major academic centers to enroll patients based on detailed molecular characteristics rather than diagnosis, and require the necessary infrastructure to analyze the high throughput sequencing data in a clinically actionable time frame [42]. Incorporation of genomics into clinical trials would only be the first step towards a more personalized therapy for AML in which each patient’s therapy is tailored to their specific molecular abnormalities. Such an approach may be able to reduce the side effects of treatment that currently are inherent in the treatment approach of acute myeloid leukemia, and enable the treatment of patients who currently are not candidates for therapy due to co-morbidities or advanced age as is the case for many patients with AML.
References
Fernandez HF, et al. Anthracycline dose intensification in acute myeloid leukemia. New Engl J Med. 2009;361:1249–59.
Döhner H, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Sanz MA, Lo-Coco F. Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol. 2011;29:495–503.
Sanz MA, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–91.
Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. New Engl J Med. 2010;363:2424–33.
Delhommeau F, et al. Mutation in TET2 in myeloid cancers. New Engl J Med. 2009;360:2289–301.
Marcucci G, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–55.
Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. New Engl J Med. 2009;361:1058–66.
Appelbaum FR, et al. Age and acute myeloid leukemia. Blood. 2006;107:3481–5.
Medeiros BC, Othus M, Fang M, Roulston D, Appelbaum FR. Prognostic impact of monosomal karyotype in young adult and elderly acute myeloid leukemia: the Southwest Oncology Group (SWOG) experience. Blood. 2010;116:2224–8.
Richardson PG, et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood. 2002;100:3063–7.
Kotla V, et al. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36.
Fehniger TA, et al. A phase 2 study of high-dose lenalidomide as initial therapy for older patients with acute myeloid leukemia. Blood. 2011;117:1828–33.
Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2012:1–10. doi:10.1038/nm.2305.
Esteller M. Epigenetics in cancer. New Engl J Med. 2008;358:1148–59.
Baylin SB. Resistance, epigenetics and the cancer ecosystem. Nat Med. 2011;17:288–9.
Sudan N, et al. Treatment of acute myelogenous leukemia with outpatient azacitidine. Cancer. 2006;107:1839–43.
Blum W, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010;107:7473–8.
Fenaux P, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28:562–9.
Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28:556–61.
Scandura JM, et al. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood. 2011;118:1472–80.
Pollyea DA, Kohrt HE, Medeiros BC. Acute myeloid leukaemia in the elderly: a review. Br J Haematol. 2011;152:524–42.
Garcia-Manero G, Stoltz ML, Ward MR, Kantarjian H, Sharma S. A pilot pharmacokinetic study of oral azacitidine. Leukemia. 2008;22:1680–4.
Garcia-Manero G, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29:2521–7.
Piekarz RL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117:5827–34.
Olsen EA, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15.
Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68.
Gore SD, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9.
Sweet MJ, Shakespear MR, Kamal NA, Fairlie DP. HDAC inhibitors: modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol Cell Biol. 2012;90:14–22.
Garcia-Manero G, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol. 2012. doi:10.1200/JCO.2011.38.3265.
Emanuele MJ, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–74.
Swords RT, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–800.
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72.
Lancet JE, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24:699–705.
Tardi P, et al. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33:129–39.
Feldman EJ, et al. First-in-man study of CPX-351: a liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J Clin Oncol. 2011;29:979–85.
Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812.
Oke A, et al. AZD1152 rapidly and negatively affects the growth and survival of human acute myeloid leukemia cells in vitro and in vivo. Cancer Res. 2009;69:4150–8.
Lowenberg B, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011;118:6030–6.
Ho J, et al. Barriers in phase I cancer clinical trials referrals and enrollment: five-year experience at the Princess Margaret Hospital. BMC Cancer. 2006;6:263.
Smith CC, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;1–6. doi:10.1038/nature11016.
Roychowdhury S, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Ungewickell, A., Medeiros, B.C. Novel agents in acute myeloid leukemia. Int J Hematol 96, 178–185 (2012). https://doi.org/10.1007/s12185-012-1151-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1151-5