
Abstract
Caspase-9 is the protease that mediates the intrinsic pathway of apoptosis, a type of cell death. Activation of caspase-9 is a multi-step process that requires dATP or ATP and involves at least two proteins, cytochrome c and Apaf-1. In this study, we mathematically model caspase-9 activation by using a system of ordinary differential equations (an ODE model) generated by a systems biology tool Simpathica—a simulation and reasoning system, developed to study biological pathways. A rudimentary version of “model checking” based on comparing simulation data with that obtained from a recombinant system of caspase-9 activation, provided several new insights into regulation of this protease. The model predicts that the activation begins with binding of dATP to Apaf-1, which initiates the interaction between Apaf-1 and cytochrome c, thus forming a complex that oligomerizes into an active caspase-9 holoenzyme via a linear binding model with cooperative interaction rather than through network formation.
Similar content being viewed by others


Introduction
Systems biology aims to understand the fundamental principles governing biological systems within a mathematical framework. There are many advantages to using a mathematical framework—the most important being that it would then be relatively easy to translate biological systems into in silico models that can be manipulated computationally and symbolically in a manner that is often simply impossible with any in vitro or in vivo models. Furthermore, it opens up the opportunities to adapt many algorithmic and analysis techniques originally devised to study natural or engineered dynamical systems that are often represented as discrete, continuous or hybrid models and analyzed using classical or modal logical frameworks.
In engineering sciences, such a consilience of model building and model checking approaches has resulted in robust and well validated engineered systems despite the fact that their design could often evolve uncontrollably in complexity to become too incomprehensible too quickly (e.g., internet or power-grid), even to the team of engineers who may have originally designed the system. But the biological systems bring up even further obstacles—often we do not know all the components participating in the process, nor possess the absolute knowledge of all the underlying interactions, complete with their kinetic parameters. To overcome these obstacles, it is important to develop system tools capable of establishing models with symbolic and logical-inference methods, analyzing them by mathematical approaches, evaluating the models with numerical simulations, and suggesting the best model system that recapitulates the biological system of interest. Recently, our group developed an automated software platform, dubbed Simpathica Model Checker, which provides “in one package” a set of the necessary systems biology tools to carry out studies of enzymatic metabolic pathways (Mishra et al. 2005). In this study we showcase an application of this platform to tease out from small number of experimental data, many of the details of biochemical processes that are thought to be occurring during apoptosis. While the study, highlighted here, is arguably limited by the data-size, it provides many insights on the nature of the “biological model checking” and the algorithmic features that systems biology tools need to incorporate.
Apoptosis is a type of cell death that removes malfunctioning or damaged cell (Evan and Littlewood 1998; Riedl and Shi 2004). Accordingly, a failure of apoptosis can contribute to diseases, such as cancer, while uncontrolled activation of the apoptotic machinery can cause unwanted loss of cells such as that occurring in neurodegenerative diseases. Apoptosis is executed by two main pathways, intrinsic, which responds primarily to intracellular stress, such as caused by chemotherapy, or extrinsic, which is activated by agonists of so-called death receptors (Cain 2003). The proteins involved in these pathways have been characterized, but it remains unclear how these proteins interact to ensure that apoptosis is prevented until needed but then executed quickly. It is likely, however, that this regulation involves quantitative changes in concentrations and activities of these proteins.
Both pathways of apoptosis can be described as a network that regulates activation of caspases, the proteases that disassemble the cell. Caspases (cysteine aspartate-specific proteases) is a family of proteins that form the execution part of the apoptotic machinery. The extrinsic pathway activates caspase-8, while intrinsic pathway activates caspase-9; either of these proteases can activate caspase-3, which does most of cell disassembly by cleaving a set of proteins. Caspase-9 functions as a holoenzyme, in which this protease is a catalytic subunit that is regulated by an oligomer of Apaf-1, also known as the apoptosome (Jiang and Wang 2004). Apaf-1 is a 130-kDa constitutively expressed protein that includes a caspase recruitment domain (CARD), a nucleotide-binding domain and 13 WD-40 repeats (Zou et al. 1997, 1999). Apaf-1 is oligomerized into the apoptosome following binding to cytochrome c in a process that requires hydrolysis of ATP or dATP (Kim et al. 2005; Yu et al. 2005). Because of experimental difficulties involved in studying the apoptosome activation, it is uncertain what sequence of reactions leads exactly to caspase-9 activation, or even how this protease is activated.
The modeling processes leading to caspase-9 activation may help to learn how to regulate caspase-9 activation for therapeutic purposes. In particular, the modeling approach could predict the outcome in a perturbed system where the balance between cell survival and cell death is compromised. Modeling can also facilitate determination of unknown factors involving the apoptotic pathway, and provide a platform for exchanging knowledge and storing information. Finally, a model of caspase-9 activation can be eventually merged with other models modularly and hierarchically to form a functional model of the cell.
Since Varner and colleagues proposed the first mathematical model of caspase activation (Fussenegger et al. 2000), several additional details have been proposed to enhance mathematical models of apoptosis by combining other major elements thought to be involved in apoptosis (Legewie et al. 2006; Nakabayashi and Sasaki 2006; Stucki and Simon 2005). For instance, there had existed earlier models to provide frameworks analogous to ours, and account for the various interactions that can affect apoptosis; however, the present study attempts to go much further in filling in many details of the intrinsic pathway that needed a clearer and more detailed description. Moreover, there exists practically no study of mathematical modeling of detailed apoptosome formation with the sole exception being the work due to Nakabayashi and Sasaki (Nakabayashi and Sasaki 2006). Even though Nakabayashi and Sasaki had modeled apoptosome assembly with the network interaction, here, in contrast to a purely simulation study as theirs, we have aimed to combine model building with model checking using novel recombinant experimental data and automated systems biology tools.
This paper proposes a mathematical approach to study and evaluate models of caspase-9 activation; additionally, it also highlights our approach to designing a software platform that can be used even by a novice user to apply, test, extend, or modify such models. This platform is based on NYU’s Simpathica Model Checker (SMC) that is part of the VALIS software environment (Paxia et al. 2002). SMC was designed for modeling, simulation, and reasoning, and is capable of effectively manipulating large, complex, and highly detailed biochemical systems (Mishra et al. 2005). Importantly, Simpathica can generate all the necessary differential equations starting from the user-defined textual or graphical descriptions, which allows even a user who is unfamiliar with this mathematical approach to apply it effortlessly. In this system, the only required inputs are reasonable ranges of initial concentrations of individual components and related metabolic reactions with proper parameters (Mishra et al. 2005). Simpathica also allows users to modify an existing model, modularly integrate with established or hypothetical models, or search over a family of plausible models, through a simple and efficient Graphical user interface (GUI) using multi-scripting facilities of VALIS. In summary, Simpathica allows users to construct and simulate models of metabolic, regulatory, and signaling networks and then to analyze their behavior with equal ease. By focusing on a well-studied and almost completely characterized system such as intrinsic apoptosis, we hope to better understand algorithmic issues inherent to an SMC-like approach.
Below, we describe how Simpathica is used to model the intrinsic apoptosis pathway as well as to check the model by comparing model-based predictions with experimental data.
Materials and methods
Recombinant system of caspase-9 activation
To study the biochemical mechanism of caspase activation, we used a recombinant system with purified recombinants Apaf-1, cytochrome c and procasepase-9 and-3 and nucleotide. Caspase-3 activities were measured over a duration, lasting 3 min because previous study had shown that caspase 3 activities would increase linearly over a time interval from 0 to 5 min (Rodriguez and Lazebnik 1999), which was also supported by the simulation data that exhibited an identical linear increment of caspase 3 activity (Supplement Fig. 2). Note that, as a convenient reference for the readers, all variables used in this study have been summarized in the Supplement Table 1.
Apaf-1 and pro-caspase-9
All reagents for recombinant system were prepared as described (Benedict et al. 2000; Fearnhead et al. 1998; Lin 2006; Rodriguez and Lazebnik 1999; Zou et al. 1999).
Assay for caspase-9 activity
We assessed caspase-9 activity by measuring conversion of procaspase-3 to caspase-3. The assay was performed as previously described elsewhere, using DEVD-AFC as a fluorogenic substrate (Rodriguez and Lazebnik 1999). Briefly, 4 μl of the tested solution was added to 0.2 ml of the assay buffer (50 mM PIPES, 10% Glycerol, 0.1 mM EDTA, 2 mM DTT, pH7.0) containing 40 μM fluorescent substrate DEVD-AFC (Biomol, Inc). The fluorescence released by caspase-3 was measured at 37°C using a Cytofluor 4000 plate reader, and the reaction rates were calculated from the fluorescence obtained before the saturation.
Assay for cytochrome c concentration
The ELISA was performed using human cytochrome c immunoassay kit (Quantikine, DCTC0 from R&D systems). Briefly, 1:1 series diluted of the reconstituted cytochrome c standard (between 0 and 20 ng/ml) and 100 μl 293 cell extract (Rodriguez and Lazebnik 1999) were transferred to polystyrene microplate coated with monoclonal antibody against cytochrome c and incubated for 2 h at room temperature. After aspiration and rinse with wash buffer, 0.2 ml of the cytochrome c conjugate was added to each well and incubated for 2 h at room temperature. After final wash, 0.2 ml of the substrate solution was added to each well and incubated for half hour in the dark. The development was stopped by adding 50 μl of stop solution and the absorbance was compared and used to calculate the concentration of cytochrome c.
Simulation program, Simpathica
The model equations and a graphical user interface (GUI) were generated, using Simpathica Model Checker (developed by NYU Bioinformatics Group, New York University, NY). This program is publicly available at NYU Bioinformatics Group website: http://bioinformatics.nyu.edu/Software/index.shtml.
Theoretical basis for simulation
Simpathica is an advanced integrated systems biology tool for simulating and reasoning about biological processes (Mishra et al. 2005). Simpathica is able to model large, modular and hierarchical biochemical pathways without making overly simplifying assumptions. For instance, at the lowest level, Simpathica may model a typical metabolic reaction modulated by an enzyme using a classical substrate-enzyme reaction. The key parameters, in Simpathica, involving a Michaelis-Menten-like enzyme-modulated reaction would be the three constants k1, k−1 and k2 (Reaction Constants).
Simpathica follows a modular scheme, much as in S-system (Voit 1991, 2000), to provide in a convenient manner a description of a biochemical pathway as a composition of several primitive reaction modules, which can be automatically translated into a set of ODE’s with additional algebraic constraints. Simpathica and XS-system (an extension of the basic S-System, (Antoniotti et al. 2002, 2003a, b)) retains this modular structure while allowing for a far richer set of modules and constraints. The Simpathica architecture is composed of two main modules and several ancillary ones. The first main module is a graphical front end that is used to construct and simulate the networks of ODE’s that are part of the model being analyzed. Simpathica uses, among others, the SBML format (System Biology Markup Language, http://www.sbml.org) for exchanging model descriptions. The second module, XSSYS is an analysis module based on a branching time temporal logic that can be used in formulating questions about the behavior of a system, represented as a set of traces (time course data) obtained from wet-lab experiments or computer simulations.
Comparison of simulation model predictions against experimental data
In this study, our model checking approach consisted of numerical comparison between simulation (in silico) data, \( \Im_{S} (U,T) \), and experimental (in vitro) data, \( \Im^{\prime}_{V} (U,T) \), leading to a better characterization of correct apoptosis model. Based on this initial ODE model, a suitable Kripke structure can be created to check more complex modal logic queries within Simpathica (data not shown).
In general, we may assume that we use an in silico model, expressed by a system of ODE’s
where u characterizes the input values, y the output values and x, all other state variables. Thus, the input–output behavior may be expressed as
Similarly, the input–output behavior observed in the in vitro experiment is expressed as
The degree of similarities between simulated and experimental results can be measured by two functions EU(T) and ET(U), as follows
and
where
and
Note that
and
Also note that, a continuous stochastic representation for EU and ET can be given as
and
If \( \Im_{V} \left( {u,t} \right) = \Im_{S} \left( {u,t} \right) \), ∀u and ∀t, then
The mean square error (MSE) was used to measure the goodness-of-fit for comparing the model predictions to the experimental data. In this case, the model yielding lower MSE values was deemed to provide a better fit.
Mathematical modeling and numerical methods
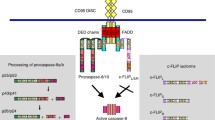
In mammalian caspase-9 pathway, the required components are Apaf-1 (A), caspase-9 (C9), cytochrome c (C), dATP (D), and caspase-3 (C3). The signaling networks regulate these components as illustrated in Fig. 1. Based on the traditional caspase-9 pathway model with a specific role for multimeric holoenzyme complex, we developed a kinetic model that was used to represent caspase-9 pathway. The differential equations, variables and default parameters used in this study are summarized in the Table 2.

Caspase-3 activation as described by a conventional diagram (a) and in Simpathica (b). Dashed line indicates enzymatic reactions. In Simpathica models, each step has its own rate constants, k p and p indicate reactions parameters, e.g., k 1 , k -1 or k 2
Modeling APAF-1, dATP and cytochrome c interactions
The first event of intrinsic apoptotic pathway is the release of cytochrome c from mitochondria after exposure to apoptotic inducers. Apaf-1 molecule binds to cytochrome c (C) and acts as the initiator for caspase activation (Cain 2003). We treat the binding of Apaf-1 and cytochrome c as reversible and the formation of complex as irreversible (Fig. 2a, c). All abbreviations for the apoptotic components are listed in Table 2.
\( \tilde{A} \bullet C \)and AC denote the intermediate molecule and Apaf-1:cytochrome c complex, respectively. Previous studies have shown that Apaf-1 also binds and hydrolyzes nucleotides such as dATP. In this reaction, cytochrome c does not affect dATP binding or hydrolysis to Apaf-1 (Zou et al. 1997). Our models treat the binding of dATP and Apaf-1:cytochrome c as reversible and the formation of complex as irreversible (Fig. 2a, c).
\( \tilde{A}\tilde{C} \bullet D \)and ACD denote the intermediate molecule and Apaf-1:cytochrome c:dATP complex (or Apaf-1 monomer), respectively. It is believed that cytochrome c binds to Apaf-1 and dATP stabilizes the binding of cytochrome c to Apaf-1 (Zou et al. 1999). Based on this information, we proposed an alternative model that Apaf-1 molecule binds to dATP first and thus, acts as the initiator for caspase activation (Fig. 2b, d).
\( \tilde{A}\tilde{D} \bullet C \) and ADC denote the intermediate molecule and Apaf-1:cytochrome c:dATP complex (or Apaf-1 monomer), respectively. We treat the binding of Apaf-1 and dATP as well as Apaf-1:dATP and cytochrome c as reversible and the formation of complex as irreversible.

Two models of initiation of caspase-9 activation and measurement of Caspase-3 activities using recombinant system and comparison with simulation data from two apoptosis initiation models. (a) and (b) are the schematic diagrams, in which the first binding molecule is Apaf-1 and (c) and (d) are the diagrams generated by the simulation program, Simpathica. We use notation 100× to indicate that rate constant of the forward reaction is increased by a scale factor of 100 (unit is M−1 s−1); similarly we denote by low affinity =10 nM−1 s−1 and high affinity =1000 nM−1 s−1. The left panels are the corresponding outputs from the model A (e and g) and the right panels are the corresponding graphs from the model B (f and h). The top panels are the corresponding outputs from the simulation with various concentration ranges of cytochrome c (e and f), and the bottom panels are the corresponding graphs from the simulation with various concentration ranges of Apaf-1 (g and h). In recombinant experiment, Caspase-3 activities were measured at seven different concentration of cytochrome c, from 0 to 120 μM and Apaf-1, from 0 to 70 nM, after 3 min (lines with closed circle). In simulations, caspase-3 activities were calculated at various concentrations of cytochrome c and Apaf-1 with two or three different a 2 (rate constant k2 in step1), i.e., 2, 20, and 2000, as well as with 10 different values of a 1 (rate constant k1 in step1) from 0.1 to 1 at increment of 0.1. The other parameters are listed in Supplement Table 1
Modeling APAF-1 multimerization
It is known that both dATP and cytochrome c are required for Apaf-1 multimerization. Cytochrome c promotes the multimerization of Apaf-1·cytochrome c complex when the dATP/ATP bound to Apaf-1 is being hydrolyzed (Zou et al. 1999). Previous studies suggest that at least seven Apaf-1 oligomers are involved, as experimentally detected and isolated using gel filtration chromatography (Zou et al. 1999). Furthermore, they also suggest that two Apaf-1 monomers form Apaf-1 dimer complex.
\( \tilde{S}_{1} \bullet \tilde{S}_{1} \) and S 2 denote the intermediate molecule and Apaf-1:cytochrome c:dATP dimer complex, respectively. We treat the binding between Apaf-1 complexes as reversible and the formation of complex as irreversible. Apaf-1 dimer complex binds Apaf-1 monomer and forms Apaf-1 trimer complex. Then, Apaf-1 dimer complex and Apaf-1 monomer form Apaf-1 trimer complex.
\( \tilde{S}_{2} \bullet \tilde{S}_{1} \) and S 2 denote the intermediate molecule and Apaf-1:cytochrome c:dATP trimer complex, respectively. We treat the binding between Apaf-1 dimer complex and Apaf-1 monomer reversible and the formation of complex as irreversible. Then, Apaf-1 trimer complex and Apaf-1 monomer combine to form Apaf-1 tetramer complex. Similar sets of equations hold for Apaf-1 pentamer, hexamer, and heptamer complexes as shown below:
During the multimerization process, if there is a cooperative interaction among Apaf-1 complexes, rate constants of binding between complexes, nth-Apaf-1 complex binding rate, γn1 will continue to increase monotonically (γ21 ≫ γ11, etc.). However, if there is no cooperative interaction among Apaf-1 complexes, rate constants of binding between complexes, γn1 will remain unaffected (γ21 = γ11, etc.).
Modeling the formation and activation of holoenzyme (or Apaf-1:cytochrome c:dATP:caspase-9 complex)
Based on previous studies, it is believed that the following sequence of events occurs: Firstly, Caspase-9 interacts with Apaf-1 only in the presence of dATP and cytochrome c; secondly, Procaspase-9 only binds to the multimeric holoenzyme complex; and finally, Apaf-1 and procaspase-9 are present at an approximately 1:1 ratio in the complex (Acehan et al. 2002; Zou et al. 1999). We made a model for binding between Apaf-1 complex and caspase-9. Apaf-1:cytochrome c:dATP:caspase-9 complex is also known as a holoenzyme.
\( \tilde{S}_{7} \bullet P9 \) and H 0 denote the intermediate molecule and inactive holoenzyme, respectively. We treat the binding between Apaf-1 heptamer complex and procaspase-9 reversible and the formation of complex as irreversible. During the binding process, if there is a cooperative interaction among procaspase-9, rate constants of binding between complexes, nth-procaspase-9 binding rate, δn 1 will be increased (δ2 1 ≫ δ1 1). However, if there is no cooperative interaction among Apaf-1 complexes, rate constants of binding between complexes, δn 1 will remain constant (δ2 1 = δ1 1). Procaspase-9 is rapidly autoactivated once bound to multimeric Apaf-1 complex.
H i and H a denote inactive holoenzyme (multimeric Apaf-1 complex with procaspase-9 or S 7:P 9) and active form (multimeric Apaf-1 complex with caspase-9 or S 7:C 9), respectively. We treat the conversion from inactive holoenzyme to active form as irreversible. Caspase-9 is fully active only when it is bound to Apaf-1 (Rodriguez and Lazebnik 1999). The activated caspase-9 is released from Apaf-1 complex.
It is assumed that new procaspase-9 has the ability to occupy the empty space in the complex to be processed.
Modeling dual roles of cytochrome c
Once Apaf-1 oligomer is formed, cytochrome c may not be required for next reaction such as caspase-9 binding and activation (Rodriguez and Lazebnik 1999). However, cytochrome c is still detected in the functional caspase-9-activating complex, which suggests that cytochrome c may have additional role in this pathway (Zou et al. 1999). Therefore, we modeled possible dual roles of cytochrome c in this study. First role is to bind Apaf-1 with dATP consistent with the assumptions of previous studies (Eqs. 13, 14). Then, we assumed that cytochrome c has another possible role, which involves activation of holoenzyme, and incorporated this assumption into the model.
C denotes cytochrome c. We treat the conversion from inactive holoenzyme to active form as irreversible.
Modeling caspase-3 activation
Procaspase-3 is hydrolyzed by active Apaf-1 holoenzyme complex (H a ).
\( \tilde{P}3 \) and C3 denote intermediate molecule and caspase-3, respectively. We treat the conversion of procaspase-3 into the intermediate molecule as reversible and the formation of active caspase-3, irreversible.
Modeling DEVD-Afc activity
DEVD-Afc is hydrolyzed by free caspase-3.
\( \tilde{V} \) and F denote intermediate molecule and fluorescent molecule Afc, respectively. We treat the conversion of DEVD-Afc into the intermediate molecule as reversible and the formation of active caspase-3, irreversible. In this study, we calculate Afc “rate” (fc/min) instead of free “Afc” concentration (nM).
The Robustness of the models
In this study, we have focused on estimating the relative model robustness among several competing network models, and used this to reconstruct the most plausible model for a biochemical pathway. We believe that this approach builds upon one of the best metrics for estimating the reliability of computationally simulated models, especially in the context where one faces relatively small-sample (and/or non-stationary) experimental data. We have tested the robustness of the models, both extensively and exhaustively, by varying the rate constant values, since most metabolic reactions, e.g., those appearing in apoptosis process, are primarily dependent on k1/k-1 ratios. In our perturbation study, we conducted our parameter-sweep by both increasing and decreasing k1 values and then simulating the models for each case to evaluate the model robustness.
Results and discussion
This study applied Simpathica, a novel systems biology platform, to model and simulate the distal part of the intrinsic pathway of apoptosis, which includes activation of caspases-9 and 3. We also compared the results of simulations to experimental data, which allowed us to optimize the models and make some conclusions regarding mechanisms of apoptosis.
Model derivation and parameter optimization
Our initial modeling is based on the current hypothesis of intrinsic apoptosis (Jiang and Wang 2000; Kim et al. 2005; Li et al. 1997; Rodriguez and Lazebnik 1999; Zheng and Flavell 2000), which is initiated by the release of cytochrome c from mitochondria (Jiang and Wang 2004). In this model (Fig. 1a), the released cytochrome c binds Apaf-1 monomer and the binding makes Apaf-1 active (step 1 in Table 1 and Fig. 1a, Eq. 13). The active Apaf-1 then binds to dATP and forms an oligomer, usually referred to as apoptosome (step 2, Eq. 14). The oligomerization is simplified in a classical enzyme reaction as reported (Nakabayashi and Sasaki 2006, step 2) or in a series of enzyme reactions (Eq. 17–22). The resulting apoptosome recruits pro-caspase-9 to form the caspase-9 holoenzyme (Eq. 23). The holoenzyme becomes active (Eq. 24, step 4) and can catalyze the activation of caspase-3 (step 5), whose activity is monitored by the increasing concentration of processed peptide substrate DEVD-Afc (step 6). The active holoenzyme can also dissociate into the apoptosome and free processed caspase-9 (Eq. 25, step 7).
Since any modeling requires quantitative parameters of modeled processes, we assembled published kinetic parameters of processes involved in caspase-9 activation, determined these parameters experimentally using an in vitro or a recombinant system, or postulated them (Table 1). We then compared the results of simulation with the published experimental data (Rodriguez and Lazebnik 1999), and changed the kinetic constants used in the simulation to reconcile the differences between the simulated and experimentally observed processes. We note that even though we checked the models of apoptosis by adopting the parameters from the literature, which were then validated by recombinant systems, it still remained necessary to check the robustness of the selected models, because model parameters are known to depend on environmental conditions such as buffer concentrations. To test the robustness of the model, various ranges of all parameters were simulated in the model system and were used to calculate the differences with experimental datasets by using serially measured mean square errors (MSE). We observed that in almost all cases parameter values selected from published kinetic reactions led to excellent fit with the most “plausible model,” i.e., the one characterized by the minimum MSE points (Supplement Fig. 1), thus indicating an extremely low likelihood of overfitting.
Remarkably, changing most kinetic parameters in the simulation, even by several orders of magnitude, had little effect on caspase-3 activation. However, changing some parameters, of critical significance, within a relatively narrow range significantly changed the kinetics of active caspase. Changing the K m and k 2 of the active holoenzyme (step 5) had the largest effect on caspase-3 activation (Supplement Fig. 1f), suggesting that these parameters are the key determinants of caspase-3 regulation. For example, decreasing the K m of the active holoenzyme increases the rate of pro-caspase-3 processing. On the other hand, decreasing the dissociation rate constant of the active holoenzyme (step 7) accelerates the cleavage and activation of both caspase-3 and caspase-9 (Supplement Fig. 1e).
Using the optimized simulation, we posed several specific questions regarding intrinsic apoptosis pathway.
1. Of the two molecules, cytochrome c or dATP, which one initiates the formation of apoptosome?
The current model of Apaf-1 activation assumes that Apaf-1 is bound to dATP, which is hydrolyzed upon binding of Apaf-1 to cytochrome c. The resulting dADP remains bound to the Apaf-1-cytochrome c complex and then is exchanged by an undefined mechanism for a molecule of dATP, thus producing a complex that is oligomerized into the apoptosome. Therefore, in this model the binding of cytochrome c to Apaf-1 is the triggering event for the formation of apoptosome and the consequent caspase-9 activation (Kim et al. 2005; Yu et al. 2005).
However, previous studies provided evidence that cytochrome c binds Apaf-1 independently of the presence of free dATP (Zou et al. 1997), that binding to cytochrome c induces binding of Apaf-1 to dATP (Cain et al. 2000; Jiang and Wang 2000), and that hydrolysis of dATP by Apaf-1 is continuous and precedes binding of cytochrome c (Zou et al. 1999). The possible discrepancies between these observations and the model suggest that the exact sequence of reactions that result in the active apoptosome is yet to be established. We used our simulation to compare the effect on caspase-9 and caspase-3 activation of two possible initiating steps: binding of cytochrome c induces binding of dATP (Fig. 2a), or binding of dATP induces binding of cytochrome c (Fig. 2b).
Simulation of the “CytC-bind-first” model showed no significant changes of caspase-3 activity in lower concentration of both cytochrome c and Apaf-1 (Fig. 2e, g). To test the robustness of the models, we also conducted extensive perturbation analysis by simulating the model with perturbed rate constants for the binding reaction of cytochrome c with Apaf-1. In this model, decreased a 1 also causes no significant changes of caspase-3 activity in lower concentration of cytochrome c and Apaf-1 (Fig. 2e). This result indicates that the formation of holoenzyme in this model is relatively independent with respect to the concentration of cytochrome c and Apaf-1. Thus, any change of cytochrome c and Apaf-1 concentration could not affect the intrinsic apoptotic system, which contradicts the experimental finding that elevation of cytochrome c induces apoptosis. However, the simulation of the second model, “ATP-bind-first” model, produced a stable increase of caspase-3 activity at even lower concentration range of cytochrome c and Apaf-1 regardless of changing rate constants. This increased activity recapitulated the shape assumed by the experimentally observed data in the recombinant system (Fig. 2f, h). Therefore, the simulation favored the model in which the first step of apoptosome formation is binding of dATP to Apaf-1—a process that then facilitates subsequent binding of cytochrome c.
2. Does cytochrome c affect caspase-9 activation after the apoptosome is formed?
Cytochrome c triggers oligomerization of Apaf-1, but is still detected in the holoenzyme, which raises the following question: Is this protein redundant in the apoptosome or does it have some specific but as-yet unidentified functions? Therefore, we compared two simulations. In one, cytochrome c is involved only in the formation of Apaf-1 complex, while in the other cytochrome c is further assumed to participate in holoenzyme activation, accompanied by increasing reaction rate. We observed that there is detectable difference between the two models. Experimental data showed that caspase-3 activity reaches its maximum value at very small amount of cytochrome c (40 μM). Simulation result clearly showed that if cytochrome c is involved in holoenzyme activation, caspase-3 activity could not reach its maximum value at even highest concentration (more than 120 μM) of cytochrome c (Fig. 3a, c). To test whether this result correlates with other components, Afc production rate is measured under a range of concentrations of Apaf-1 (Fig. 3d, e). This result is found consistent with the result from previous simulation, and thus, prompting us to conclude that cytochrome c is unlikely to be involved in the caspase-9 activation. However, this analysis does not exclude the possibility that cytochrome c in the complex may still have a role in stabilizing the holoenzyme complex. We further tested a seemingly plausible hypothesis that slowing down the reaction rate could lead the simulation study to a different conclusion, thus putting in doubt the significance of the single-role model. The simulation data showed that reaction rate could be suitably slowed down in the simulation to find a rate that fits the unfavorable dual-role model the best, and brings it as close as possible to the single-role model. Nonetheless, the mean square error test continued to support the single-role model (Supplement Fig. 3) over the best dual-role model.

Test to determine a second role of cytochrome c in intrinsic apoptotic pathway besides the formation of Apaf-1 oligomer. The Diagram shows the modeling pathway view in Simpathica (a). Bold arrow in the diagram indicates that cytochrome c is involved in Apaf-1 complex formation. Broken arrow in the diagram indicates that cytochrome c is also involved in another role, activating holoenzyme. Two models, one assigning a single role (b and d) to cytochrome c and the other assigning a dual role (c and e), were tested in two components, cytochrome c (b and c) and Apaf-1 (d and e). The short arrow in (b) indicates the concentration of cytochrome c (40 μM) at which caspase-3 activity reaches the maximum activity in a graph corresponding to the model in which cytochrome c is only involved in the formation of Apaf-1 complex. The short arrow in (c) indicates the concentration of cytochrome c (more than 120 μM) at which caspase-3 activity reaches its peak value in a graph corresponding to the model in which cytochrome c has a dual role, controlling how Apaf-1 complex (arrow with solid line in the left diagram) and enzymatic activity facilitates holoenzyme activation (arrow with broken line in the left diagram). Caspase-3 activities were measured at seven different concentration of cytochrome c, from 0 to 120 μM after 3 min. The complete set of parameters is listed in the Supplement Table 1
3. Is there positive cooperative interaction during the formation of Apaf-1 complex?
A key step in activation of caspase-9 is oligomerization of Apaf-1. At present, how this oligomerization occurs is not fully understood. The rapid rate of the oligomerization and the number of the subunits in the oligomer suggests a possibility of cooperative binding among the subunits. Recently, Nakabayashi and Sasaki mathematically modeled apoptosome assembly with a quadratic network interaction framework that did not involve cooperative binding (Nakabayashi and Sasaki 2006).
To test whether cooperative binding can also explain how Apaf-1 is oligomerized, we simulated the effect of cooperative binding on the activation of caspases-9 and -3, we assumed that Apaf-1 oligomerization proceeds stepwise (Eqs (17) through (22)). We created two simulation models with a linear network interaction framework. In one of the models, each Apaf-1 complex subunit has the same binding rate as previously bound subunits (γ11 = γ21 = γ31 ··· = γ71; γn 1 is binding rate of nth Apaf-1 complex), while in the other model the successive Apaf-1 complex subunits have monotonically increasing binding rates (γ11 ≪ γ21 ≪ γ31 ··· γ71).
For comparison, we also simulated the Nakabayashi and Sasaki’s complex network model (Nakabayashi and Sasaki 2006) in which each Apaf-1 monomer binding to the oligomer has the same binding rate, but the monomers and oligomers participate in complex network interactions. For example, an Apaf-1 trimer can bind to a tetramer to form the heptameric apoptosome, or bind to a dimer, to form an intermediate pentamer, and so on. In this model, a total of 21 (=(7 × 6)/2) individual interactions would be necessary to connect all components needed in the formation of a heptamer (Fig. 4c).

Cooperative binding of Apaf-1 can explain the rate of caspase-3 activation. Top panel shows the modeling pathways view in Simpathica and bottom panel shows the results from simulation and in vitro experiment. a A linear network model with non-cooperative interactions. b A linear network model with cooperative interactions. c A complex network model with non-cooperative interactions. Dashed lines indicate graphs from the recombinant experiment. Caspase-3 activities were measured at seven different concentration of Apaf-1, from 0 to 70 nM. Caspase-3 activities were calculated at various concentration of Apaf-1 up to 70 nM (red lines). The complete set of parameters is listed in Supplement Table 1
In the linear model the increase of caspase-3 activity was exponential at low rate constant and weakly sigmoidal at higher rate constant if no positive cooperativity was assumed (Fig. 4a), and sigmoidal if positive cooperativity was introduced (Fig. 4b). The Nakabayashi and Sasaki model produced a sigmoidal curve that was initially nearly linear (Fig. 4c). To determine which of the curves fits better the experimental results, we calculated the mean square error (MSE, see Theoretical basis for Simulation) between experimental and simulation data using a least square distance function, and concluded that the model based on a linear network with cooperative interactions (Fig. 4b) fits the data the best. We also tested the robustness of the models by varying the binding rate constants. As we expected, increased binding rates elevate the holoenzyme activity. However, the resulting exponential input–output curve at low rate constant remained unchanged and overall shape remained more or less the same. These simulation results indicate that modified parameters do not change the patterns of the dynamics. Therefore, we may reasonably conclude that there exist positive cooperative interactions during the Apaf-1 complex formation.
4. Is binding of procapase-9 to the apoptosome cooperative?
Caspase-9 functions as a holoenzyme in which this protease and Apaf-1 are present in 1:1 ratio (Acehan et al. 2002). Thus, total of seven caspase-9 molecules can exist in holoenzyme complex even though it is not clear how this oligomerization occurs. The simulation suggesting a positive cooperative binding among Apaf-1 complex subunits (Fig. 4) led us to test whether binding of procaspase-9 to the oligomer is also cooperative. We compared two alternative models. In one model, each procaspase-9 molecule binds with the same binding constant as the previous procaspase-9 during the formation of the holoenzyme (δ11 = δ21 = δ31 ··· = δ71; δn 1 is binding rate of nth procaspase-9 molecule), while in the other alternative model the rate of binding of each successive procaspase-9 molecules increases monotonically (δ11 ≪ δ21 ≪ δ31 … ≪ δ71).
We found that assuming positive cooperative binding of caspase-9 to the Apaf-1 oligomer could not by itself explain the sigmoidal shape in the rate of caspase-3 activation observed experimentally (Fig. 5 left and right panel). Moreover, positive cooperative binding of caspase-9 to the Apaf-1 oligomer could accelerate the holoenzyme activity in the higher concentration of Apaf-1 relative to the activity measured by the experimental data. We also tested the robustness of the models by perturbing binding rate constants. However, since the overall shape remained the same, we concluded that caspase-3 activation involves positive cooperative interaction among Apaf-1 molecules during the formation of Apaf-1 multimeric complex in low concentration of Apaf-1 (Fig. 5 bottom panel).

Comparison of cooperative interactions among procaspase-9 during the formation of holoenzyme complex and cooperative interaction among Apaf-1 complex subunits during the formation of Apaf-1 multimeric complex. Left panel exhibits an absence of cooperative interaction among procaspase-9 during the formation of holoenzyme complex. Right panel exhibits a cooperative interaction among procaspase-9 during the formation of holoenzyme complex. δ1 indicates rate constant for the binding of procaspase-9. Top panel exhibits no cooperative interaction among Apaf-1 complex subunits during the formation of Apaf-1 multimeric complex. Bottom panel exhibits a cooperative interaction among Apaf-1 complex subunits during the formation of Apaf-1 multimeric complex γ1 (rate constant k1 in step 2-1) indicates rate constant for the binding of Apaf-1 complex (unit of γ and δ is M−1 s−1). In all simulation, caspase-3 activities were calculated at various concentration of Apaf-1 up to 70 nM. Bold lines indicate graphs from the recombinant experiment. Caspase-3 activities were measured at seven different concentration of Apaf-1, from 0 to 70 nM. A complete list of parameters appears in Supplement Table 1
Based on the previous results, we proposed that seven Apaf-1 complex subunits interact cooperatively with each other while forming the apoptosome, and that this positive cooperation can explain the rapidity of caspase-9 activation during apoptosis, especially considering that assuming positive cooperativity in the binding of caspase-9 to the apoptosome failed to affect the rates of caspase-3 activation
5. Is free caspase-9 active?
The experimental evidence indicates that caspase-9 is fully active only when it is bound to Apaf-1 (Rodriguez and Lazebnik 1999; Stennicke et al. 1999), while earlier studies suggested that processed caspase-9 can also be active (Cain et al. 2000; Zou et al. 1999). We simulated these two possibilities (Fig. 6a, b) and found that the first model fits the experimental findings better (Fig. 6c, e). Simulation study shows that if free caspase-9 also has any activity, holoenzyme activity in high concentration of Apaf-1 should have been much higher than what we observed in experiments. When we compared the activity of free caspase-9 against that of complex-bound caspase-9s, we observed that holoenzyme activity was increased by almost 50%, in higher concentration of Apaf-1. However, if we examine the activities at a low level of free or complex-bounded caspase-9s, we observed both holoenzyme activities to be of similar magnitudes. Therefore, we concluded that free caspase-9 has none or very little influence. Our simulation data confirmed the result from recent experimental data that caspase-9 is fully active only when it is bound to Apaf-1 (Rodriguez and Lazebnik 1999), and that, in contradiction to earlier mathematical models, the apoptotic process is maintained primarily by caspase-3 or caspase-9 (Fussenegger et al. 2000; Legewie et al. 2006; Stucki and Simon 2005).

Holoenzyme-bound caspase-9 is enough to activate caspase-3. Panel (a) shows the schematic diagrams, which implicitly assume a role only for holoenzyme-bound caspase-9 in activating caspase-3. Panel (b) shows the schematic diagrams, which assume that both holoenzyme bound caspase-9 and free caspase-9 jointly activate caspase-3. Bottom panels (c to f) are the corresponding graphs from the simulation with various concentration ranges of cytochrome c (g and h) and Apaf-1 (e and f). The left panels (c and e) are the outputs of the simulation model from panel A and the right panels (d and f) are the outputs of a simulation model from panel B. In recombinant experiment as well as simulation, caspase-3 activities were measured at similar concentrations described in Fig. 2. A complete list of parameters appears in Supplement Table 1
Based on simulation data, we reconstructed the model for the intrinsic apoptotic pathway (Fig. 7). Based on the analysis from this study, we concluded that the best target molecule to control apoptosis is not necessarily initiator caspase such as caspase-9 but a holoenzyme complex. Although the behavior of caspase-3 activity is mainly caused by positive cooperative binding of Apaf-1 subunits, cooperative binding of procaspase-9 still can affect caspase-3 activity in low concentration of Apaf-1. It is interesting that the simulation data from this study based on positive cooperative interactions almost matched exactly with in vitro experimental data for all data points. This result supports the main claim of this study that all components and their interactions used in this simulation study are structured appropriately in their ability to use the experimental data to distinguish among a family of plausible models.

Revised caspase-9 dependent intrinsic apoptotic pathway. Variable γ denotes rate constant for the binding between Apaf-1 complex units during the formation of apoptosome and δ denotes rate constant for the binding between multimeric Apaf-1 complex and procaspase-9
References
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW (2002) Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9:423–432
Antoniotti M, Policriti A, Ugel N, Mishra B (2002) XS-systems: extended S-systems and algebraic differential automata for modeling cellular behaviour. In: Sahni S, Prasanna VK, Shukla U (eds) Proceeding of HiPC, LNCS. 2552:431–442
Antoniotti M, Park F, Policriti A, Ugel N, Mishra B (2003a) Foundations of a query and simulation system for the modeling of biochemical and biological processes. Pac Symp Biocomput, pp 116–127
Antoniotti M, Piazza C, Policriti A, Sineoni M, Mishra B (2003b) Modeling cellular behavior with hybrid automata: Bisimulation and collapsing. In: Priami C (ed) International workshop on computational methods in systems biology, LNCS. 2602:57–74
Benedict MA, Hu Y, Inohara N, Nunez G (2000) Expression and functional analysis of Apaf-1 isoforms, Extra Wd-40 repeat is required for cytochrome c binding and regulated activation of procaspase-9. J Biol Chem 275:8461–8468
Cain K (2003) Chemical-induced apoptosis: formation of the Apaf-1 apoptosome. Drug Metab Rev 35:337–363
Cain K, Bratton SB, Langlais C, Walker G, Brown DG, Sun XM, Cohen GM (2000) Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J Biol Chem 275:6067–6070
Evan G, Littlewood T (1998) A matter of life and cell death. Science 281:1317–1322
Fearnhead HO, Rodriguez J, Govek EE, Guo W, Kobayashi R, Hannon G, Lazebnik YA (1998) Oncogene-dependent apoptosis is mediated by caspase-9. Proc Natl Acad Sci USA 95:13664–13669
Fussenegger M, Bailey JE, Varner J (2000) A mathematical model of caspase function in apoptosis. Nat Biotechnol 18:768–774
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273:32608–32613
Jiang X, Wang X (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J Biol Chem 275:31199–31203
Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73:87–106
Kim HE, Du F, Fang M, Wang X (2005) Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci USA 102:17545–17550
Legewie S, Bluthgen N, Herzel H (2006) Mathematical modeling identifies inhibitors of apoptosis as mediators of positive feedback and bistability. PLoS Comput Biol 2:1061–1073
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489
Lin S (2006) Regulation of caspase-9 holoenzyme. Dissertation, Stony Brook University
Mishra B, Antoniotti M, Paxia S, Ugel N (2005) Simpathica: a computational systems biology tool within the valis Bioinformatics environment. In: Eiles E, Kriete A (eds) Computational systems biology. Elsevier, Amsterdam
Moretti A, Weig HJ, Ott T, Seyfarth M, Holthoff HP, Grewe D, Gillitzer A, Bott-Flugel L, Schomig A, Ungerer M, Laugwitz KL (2002) Essential myosin light chain as a target for caspase-3 in failing myocardium. Proc Natl Acad Sci USA 99:11860–11865
Nakabayashi J, Sasaki A (2006) A mathematical model for apoptosome assembly: the optimal cytochrome c/Apaf-1 ratio. J Theor Biol 242:280–287
Paxia S, Rudra A, Zhou Y, Mishra B (2002) A random walk down the genomes: DNA evolution in VALIS. Computer 35(7):73–79
Purring C, Zou H, Wang X, McLendon G (1999) Stoichiometry, free energy and kinetic aspects of cytochrome c: Apaf-1 binding in apoptosis. J Am Chem Soc 121:7435–7436
Purring-Koch C, McLendon G (2000) Cytochrome c binding to Apaf-1: the effects of dATP and ionic strength. Proc Natl Acad Sci USA 97:11928–11931
Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5:897–907
Rodriguez J, Lazebnik Y (1999) Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 13:3179–3184
Ryan CA, Stennicke HR, Nava VE, Burch JB, Hardwick JM, Salvesen GS (2002) Inhibitor specificity of recombinant and endogenous caspase-9. Biochem J 366:595–601
Skoog L, Bjursell G (1974) Nuclear and cytoplasmic pools of deoxyribonucleoside triphosphates in Chinese hamster ovary cells. J Biol Chem 249:6434–6438
Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS (1998) Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 273:27084–27090
Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS (1999) Caspase-9 can be activated without proteolytic processing. J Biol Chem 274:8359–8362
Stucki JW, Simon HU (2005) Mathematical modeling of the regulation of caspase-3 activation and degradation. J Theor Biol 234:123–131
Voit EO (1991) Canonical nonlinear modeling: S-system approach to understanding complexity. Van Nostrand Reinhold, New York
Voit EO (2000) Computational analysis of biochemical systems: a practical guide for biochemists and molecular biologists. Cambridge University Press, New York
Yu X, Acehan D, Menetret JF, Booth CR, Ludtke SJ, Riedl SJ, Shi Y, Wang X, Akey CW (2005) A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure 13:1725–1735
Zheng TS, Flavell RA (2000) Death by numbers. Nat Biotechnol 18:717–718
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90:405–413
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556
Zubay G (1993) Biochemistry, 3rd edn. Wm. C. Brown Publishers, Oxford
Acknowledgment
We thank Dr. Gabriel Nunez (University of Michigan) for providing Apaf-1 cDNA.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Seongho Ryu and Shih-chieh Lin contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ryu, S., Lin, Sc., Ugel, N. et al. Mathematical modeling of the formation of apoptosome in intrinsic pathway of apoptosis. Syst Synth Biol 2, 49–66 (2008). https://doi.org/10.1007/s11693-009-9022-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11693-009-9022-y