
ABSTRACT
Purpose
Chlorogenic acid (CGA), the most abundant component in coffee, has exhibited many biological activities. The objective of this study is to assess preventive and therapeutic effects of CGA on obesity and obesity-related liver steatosis and insulin resistance.
Methods
Two sets of experiments were conducted. In set 1, 6-week old C57BL/6 mice were fed a regular chow or high-fat diet (HFD) for 15 weeks with twice intra-peritoneal (IP) injection of CGA (100 mg/kg) or DMSO (carrier solution) per week. In set 2, obese mice (average 50 g) were treated by CGA (100 mg/kg, IP, twice weekly) or DMSO for 6 weeks. Body weight, body composition and food intake were monitored. Blood glucose, insulin and lipid levels were measured at end of the study. Hepatic lipid accumulation and glucose homeostasis were evaluated. Additionally, genes involved in lipid metabolism and inflammation were analyzed by real time PCR.
Results
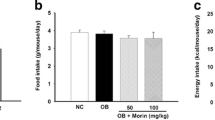
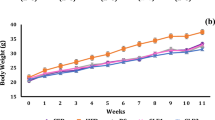
CGA significantly blocked the development of diet-induced obesity but did not affect body weight in obese mice. CGA treatment curbed HFD-induced hepatic steatosis and insulin resistance. Quantitative PCR analysis shows that CGA treatment suppressed hepatic expression of Pparγ, Cd36, Fabp4, and Mgat1 gene. CGA treatment also attenuated inflammation in the liver and white adipose tissue accompanied by a decrease in mRNA levels of macrophage marker genes including F4/80, Cd68, Cd11b, Cd11c, and Tnfα, Mcp-1 and Ccr2 encoding inflammatory proteins.
Conclusion
Our study provides direct evidence in support of CGA as a potent compound in preventing diet-induced obesity and obesity-related metabolic syndrome. Our results suggest that drinking coffee is beneficial in maintaining metabolic homeostasis when on a high fat diet.









Similar content being viewed by others

REFERENCES
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the global burden of disease study 2013. Lancet 2014;384:766-81.
Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
van Dam RM. Coffee consumption and risk of type 2 diabetes, cardiovascular diseases, and cancer. Appl Phys Nutr Metab. 2008;33:1269–83.
Olthof MR, Hollman PCH, Katan MB. Chlorogenic acid and caffeic acid are absorbed in humans. J Nutr. 2001;131:66–71.
Azuma K, Ippoushi K, Nakayama M, Ito H, Higashio H, Terao J. Absorption of chlorogenic acid and caffeic acid in rats after oral administration. J Agric Food Chem. 2000;48:5496–500.
McCarty MF. A chlorogenic acid-induced increase in GLP-1 production may mediate the impact of heavy coffee consumption on diabetes risk. Med Hypotheses. 2005;64:848–53.
Prabhakar PK, Doble M. Synergistic effect of phytochemicals in combination with hypoglycemic drugs on glucose uptake in myotubes. Phytomedicine. 2009;16:1119–26.
Ong KW, Hsu A, Tan BK. Chlorogenic acid stimulates glucose transport in skeletal muscle via AMPK activation: a contributor to the beneficial effects of coffee on diabetes. PLoS One. 2012;7:e32718.
Ong KW, Hsu A, Tan BK. Anti-diabetic and anti-lipidemic effects of chlorogenic acid are mediated by ampk activation. Biochem Pharmacol. 2013;85:1341–51.
Rodriguez de Sotillo DV, Hadley M. Chlorogenic acid modifies plasma and liver concentrations of: cholesterol, triacylglycerol, and minerals in (fa/fa) Zucker rats. J Nutr Biochem. 2002;13:717–26.
Wan CW, Wong CN, Pin WK, Wong MH, Kwok CY, Chan RY, et al. Chlorogenic acid exhibits cholesterol lowering and fatty liver attenuating properties by up-regulating the gene expression of PPAR-alpha in hypercholesterolemic rats induced with a high-cholesterol diet. Phytother Res. 2013;27:545–51.
Cho AS, Jeon SM, Kim MJ, Yeo J, Seo KI, Choi MS, et al. Chlorogenic acid exhibits anti-obesity property and improves lipid metabolism in high-fat diet-induced-obese mice. Food Chem Toxicol. 2010;48:937–43.
Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013;30:1447–57.
Perlemuter G, Bigorgne A, Cassard-Doulcier AM, Naveau S. Nonalcoholic fatty liver disease: from pathogenesis to patient care. Nat Clin Pract Endocrinol Metab. 2007;3:458–69.
Monteiro R, Azevedo I. Chronic inflammation. In Obesity and the Metabolic Syndrome. Mediators Inflamm 2010. doi:10.1155/2010/289645.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
Bu L, Gao M, Qu S, Liu D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J. 2013;15:1001–11.
Cui R, Gao M, Qu S, Liu D. Overexpression of superoxide dismutase 3 gene blocks high-fat diet-induced obesity, fatty liver and insulin resistance. Gene Ther. 2014;21:840-8.
Shan J, Fu J, Zhao Z, Kong X, Huang H, Luo L, et al. Chlorogenic acid inhibits lipopolysaccharide-induced cyclooxygenase-2 expression in RAW264.7 cells through suppressing NF-kappaB and JNK/AP-1 activation. Int Immunopharmacol. 2009;9:1042–8.
Shi HT, Dong L, Jiang J, Zhao JH, Zhao G, Dang XY, et al. Chlorogenic acid reduces liver inflammation and fibrosis through inhibition of toll-like receptor 4 signaling pathway. Toxicology. 2013;303:107–14.
Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–87.
Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–89.
Yu ST, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma 1 (PPAR gamma 1) overexpression. J Biol Chem. 2003;278:498–505.
Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–76.
Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–47.
Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, et al. Role for PPAR gamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–50.
Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–67.
Kim Y, Park T. DNA microarrays to define and search for genes associated with obesity. Biotechnol J. 2010;5:99–112.
Miquilena-Colina ME, Lima-Cabello E, Sanchez-Campos S, Garcia-Mediavilla MV, Fernandez-Bermejo M, Lozano-Rodriguez T, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60:1394–402.
Koonen DPY, Jacobs RL, Febbraio M, Young ME, Soltys CLM, Ong H, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56:2863–71.
Ma Y, Liu D. Activation of pregnane X receptor by pregnenolone 16 alpha-carbonitrile prevents high-fat diet-induced obesity in AKR/J mice. Plos One. 2012;7:e38734.
Gao M, Ma Y, Liu D. Rutin suppresses palmitic acids-triggered inflammation in macrophages and blocks high fat diet-induced obesity and fatty liver in mice. Pharm Res. 2013;30:2940–50.
Gao M, Ma Y, Cui R, Liu D. Hydrodynamic delivery of FGF21 gene alleviates obesity and fatty liver in mice fed a high-fat diet. J Control Release. 2014;185:1–11.
Cortes VA, Curtis DE, Sukumaran S, Shao XL, Parameswara V, Rashid S, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized Lipodystrophy. Cell Metab. 2009;9:165–76.
Lee YJ, Ko EH, Kim JE, Kim E, Lee H, Choi H, et al. Nuclear receptor PPAR gamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc Natl Acad Sci U S A. 2012;109:13656–61.
Huxley R, Lee CM, Barzi F, Timmermeister L, Czernichow S, Perkovic V, et al. Coffee, decaffeinated coffee, and tea consumption in relation to incident type 2 diabetes mellitus: a systematic review with meta-analysis. Arch Intern Med. 2009;169:2053–63.
Ahrens MJ, Thompson DL. Effect of emulin on blood glucose in type 2 diabetics. J Med Food. 2013;16:211–5.
Karthikesan K, Pari L, Menon VP. Protective effect of tetrahydrocurcumin and chlorogenic acid against streptozotocin-nicotinamide generated oxidative stress induced diabetes. J Funct Foods. 2010;2:134–42.
Xu HY, Barnes GT, Yang Q, Tan Q, Yang DS, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30.
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported in part by the National Institute of Health (RO1EB007357 and RO1HL098295). We thank Ms. Ryan Fugett for proof-reading and English editing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, Y., Gao, M. & Liu, D. Chlorogenic Acid Improves High Fat Diet-Induced Hepatic Steatosis and Insulin Resistance in Mice. Pharm Res 32, 1200–1209 (2015). https://doi.org/10.1007/s11095-014-1526-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-014-1526-9