
Abstract
Type I interferons (IFNs) are produced by leukocytes in reaction to pathogenic infection and function as positive mediators in antiviral pathways. Among IFNs, IFN alpha (IFNA) has the largest number of family members and plays an important role against the invasion of pathogens. Bats are putative and proven vectors for numerous viruses; however, the evolution of the IFNA family in bats has not been addressed. Here, we construct a phylogeny of IFNA families, including one fruit bat (Dobsonia viridis), with other vertebrates as references. Site-model estimation reveals that positive selection has shaped bat IFNA genes, showing that positive selection drives the evolution of bat IFNA genes.
Similar content being viewed by others


Introduction
Type I IFN homologous proteins are found in many organisms including mammalian and nonmammalian species (Schultz et al. 2004). They function to activate the IFN signaling pathways by binding to the IFNA receptors IFNAR1 and IFNAR2 (Cutrone and Langer 1997). IFNA proteins are secreted by leukocytes during the innate immune response against viral infection and stimulate cellular resistance to such infections by the induction of antiviral proteins like eukaryotic translation initiation factor 2-alpha kinase 2 (EIF2AK2), 2′,5′-oligoadenylate synthetase, RNase L, and the Mx protein GTPases (Pestka et al. 1987; Samuel 2001).
The typical type I IFN signaling pathway starts from recognition between IFN and IFN receptors (IFNAR1 and IFNAR2), followed by the JAK-STAT signaling pathway (O’Shea et al. 2002). Invasion of DNA or RNA viruses can be recognized by host pattern recognition receptors that are expressed in immune cells (Theofilopoulos et al. 2005; Kawai and Akira 2006). The type I IFNs are key cytokines that are produced after pathogenic infection and can induce both the innate immune response and the subsequent adaptive immunity to pathogens (Theofilopoulos et al. 2005; Kawai and Akira 2006). Type I IFNs provide the most effective antiviral abilities, and even a weak signal of infection could enhance a response of the immune cells (Taniguchi and Takaoka 2001). Recombinant IFNAs have been clinically approved for antiviral therapy for humans (Bekisz et al. 2004).
Bats can carry more than 60 kinds of virus, including emerging or re-emerged viruses like Ebola virus, Henipah virus, SARS-like Coronavirus, Japanese encephalitis virus, and Lyssavirus (Halpin et al. 1999; Yob et al. 2001; Leroy et al. 2005; Li et al. 2005; Calisher et al. 2006; Cui et al. 2008). It has been reported that Ebola and West Nile viruses invade host cells by blocking the activation of the interferon signaling pathway (Basler et al. 2003; Guo et al. 2005; Reid et al. 2006). Thus, IFN families play pivotal roles in the complex cytokine networks and defense against pathogens (Pestka et al. 1987; Samuel 2001). Bats may have special immunological systems (Dobson 2005; Calisher et al. 2006), thus requiring adaptive evolution of the immune system to conquer these bat pathogens.
Gene conversion and gene duplication are the main forces that drive the evolution of the mammalian type I IFN gene family (Hughes 1995; Woelk et al. 2007). Phylogenetic analysis of the evolutionary history of mammalian type I IFN suggests that IFNA genes have duplicated independently within different mammalian orders and have been subjected to purifying selection (Hughes 1995). Especially for the IFNA genes, gene conversion has become the main evolutionary force after the divergence of birds and mammals but before the divergence of the ancestors of eutherian species (Shaw et al. 1983; Gillespie and Carter 1983; Hughes 1995). The selection pressures shaping the evolution of the bat IFNA family remain unknown. Here we provide a phylogenetic analysis of the bat IFNA genes and use maximum likelihood methods to evaluate the molecular evolution in the bat IFNA family.
Materials and Methods
Cloning and Sequencing
Genomic DNA was extracted from 3-mm biopsy punches (Stiefel, Germany) of one bat (Dobsonia viridis), following the manufacturer’s protocol provided with the DNeasy Blood and Tissue Kit (Qiagen, Switzerland). We designed a pair of degenerate primers to amplify the bat IFNA genes: BATINFAF [5′-ATGGC(C/T)CTGCCCT(G/T)TTCCTT(C/A)CTGATGG-3′] and BATINFR [5′-AAGAG(A/T)AGGA(T/C)CTCATGATCTCTGCTCTGA-3′]. The N terminus primer encodes the first 28 nucleotides of the coding region. The C terminus primer, positioned at the downstream side of the coding region, is missing the last 34 nucleotides; thus, the gene products are about 530 bp. Conditions of the polymerase chain reaction (PCR) were 94°C for 5 min; 33 cycles at 94°C for 30 s, 57°C for 30 s, and 72°C for 1 min; and 72°C for 10 min. The PCR volume (50 μl) contained 50 ng genomic DNA, 10 μM each primer, and 25 μl 2× ExTaq polymerase mix (Takara, Japan). PCR products were purified using the Agarose Gel DNA Purification Kit (Takara) and cloned into the pMD19-T vector (Takara). The positive clones were then sequenced in both directions with the universal M13 primers M13-47 (5′-CGCCAGGGTTTTCCCAGTCACGAC-3′) and M13-48 (5′-GAGCGGATAACAATTTCACACAGG-3′), using a Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, USA) on an ABI 3730 sequencer (Applied Biosystems). All the new IFNA genes were submitted to NCBI under accession nos. HM217768-HM217775.
Phylogenetic Reconstruction
The phylogenetic reconstruction was conducted using the neighbor-joining method, with 1,000 bootstrap replications, in Mega 4 (Tamura et al. 2007). The evolutionary distances were computed using the maximum composite likelihood method (Tamura et al. 2004). The bat IFNA sequences and the published subtypes from cat (Felis catus), ferret (Mustela putorius furo), panda (Ailuropoda melanoleuca), pig (Sus scrofa), human (Homo sapiens), and mouse (Mus musculus) (Supplementary Table 1) were used to resolve the evolutionary history of the IFNA gene family. Four sequences of interferon omega (Supplementary Table 1) were used to root the tree. All the sequences were aligned using Clustal X (Thompson et al. 1997) based on the amino acid sequences.
Molecular Evolution
To detect gene conversion, GeneConv software (version 1.81a) was used (Sawyer 1989). To evaluate the pattern of nucleotide substitution in and among sites, the number of synonymous substitutions per synonymous site and the number of nonsynonymous substitutions per nonsynonymous site were estimated in PAML4 (Yang 2007). Two pairs of models are recommended: M1a (a neutral model characterized as 0 < ω0 < 1, ω1 = 1) versus M2a (a positive selection model characterized as ω2 >1); M7 (a β-distributed model constrained by ω <1) versus M8 (a, β, and ω model to evaluate positively selected sites) (Yang 2007). Likelihood ratio tests were also performed to test for positive selection (Yang 1998), and the Bayesian posterior probability of potential selected sites was evaluated under Bayes empirical Bayes (BEB) criteria.
Results
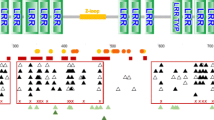
We sequenced 81 positive clones of Dobsonia viridis and successfully obtained eight IFNA gene types (with amino acid similarity 88.4–99.4%) and one pseudogene. Our phylogenetic tree based on all IFNA genes (without the pseudogene) exhibited moderately well-grouped clades of the superorder Laurasiatheria (bat, cow, cat, panda, and ferret; bootstrap value 60) and the superorder Euarchontoglires (human and mouse; bootstrap value 70) (Fig. 1).

Neighbor-joining tree of IFNA gene families. The tree is rooted with interferon omega genes, and the maximum composite likelihood method is used. Percentage bootstrap values (higher than 50) are shown on interior branches
In the output, GeneConv identified that gene conversion could happen between a pair of sequences (IFNA7 and 8) from the alignment (Supplementary Table 2). Both models allowing sites under positive selection (M2a and M8) showed significant differences against the neutral models (M1a and M7) in likelihood ratio tests (Table 1). The signal for positive selection was decreased when IFNA7, which was shown to undergo positive selection, was removed, and the ω ratio was also lower (3.488 vs. 2.697). Thus, our results also suggest that consideration of gene conversion is necessary for evaluation of selective pressure among the gene family.
Discussion
Duplication is the main force leading to the evolution of gene families (Ohno et al. 1968), and positive selection of a gene family could result in diversification among gene family members, which suggests that the divergent regions may be related to specific functions of family members (Hughes and Hughes 1993; Hughes 1994). An elevated ω ratio (>1) is an indicator of positive selection among gene families (Hughes 2000). Our results revealed that positive selection had favored the bat INFA genes with the detection of positively selected sites among bat IFNA genes, and gene conversion could contribute to a change of selective force. Among the four amino acid sites under positive selection (after removing IFNA7), positions 11 and 15 belong to the signal peptide region that directs protein transport to certain organelles, such as the endoplasmic reticulum, and codons 54 and 107 belong to the chain region. Of all type I IFNs, IFNA has the largest number of subtypes (Shaw et al. 1983; Pestka et al. 2004), and all subtypes may have different biological activities (Foster et al. 1996; Yeow et al. 1998; Baldwin et al. 2004). We have also successfully cloned the multiple genotypes of bat IFNA, and our evolutionary analysis suggested that evolution of the bat IFNA family has been favored by positive selection. The cloned pseudogene revealed that the evolutionary loss of function also shaped the bat genome during IFNA evolution.
Pathogens have evolved various ways to avoid the antiviral effects of the host’s immune system. Type I IFN, however, induces the most basic defense mechanisms against those pathogens (García-Sastre and Biron 2006). The positive selection noted in bat IFNA genes probably reflects the conflict between bat pathogens and the interferon-mediated immune system, as the pathogens evolve blocks to recognition by the immune system and the immune system tries to maintain recognition. This phenomenon is probably not unique to bats, and drawing conclusions about it would require positive selection analysis of IFNA genes in a broad range of other species.
References
Baldwin SL, Powell TD, Sellins KS, Radecki SV, Cohen JJ, Milhausen MJ (2004) The biological effects of five feline IFN-alpha subtypes. Vet Immunol Immunopathol 99(3–4):153–167
Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Mühlberger E, Bray M, Klenk HD, Palese P, García-Sastre A (2003) The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77(14):7945–7956
Bekisz J, Schmeisser H, Hernandez J, Goldman ND, Zoon KC (2004) Human interferons alpha, beta and omega. Growth Factors 22(4):243–251
Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T (2006) Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev 19(3):531–545
Cui J, Counor D, Shen D, Sun G, He H, Deubel V, Zhang S (2008) Detection of Japanese encephalitis virus antibodies in bats in Southern China. Am J Trop Med Hyg 78(6):1007–1011
Cutrone EC, Langer JA (1997) Contributions of cloned type I interferon receptor subunits to differential ligand binding. FEBS Lett 404(2–3):197–202
Dobson AP (2005) What links bats to emerging infectious diseases? Science 310(5748):628–629
Foster GR, Rodrigues O, Ghouze F, Schulte-Frohlinde E, Testa D, Liao MJ, Stark GR, Leadbeater L, Thomas HC (1996) Different relative activities of human cell-derived interferon-alpha subtypes: IFN-alpha 8 has very high antiviral potency. J Interferon Cytokine Res 16(12):1027–1033
García-Sastre A, Biron CA (2006) Type I interferons and the virus-host relationship: a lesson in détente. Science 312(5775):879–882
Gillespie D, Carter W (1983) Concerted evolution of human interferon alpha genes. J Interferon Res 3(1):83–88
Guo JT, Hayashi J, Seeger C (2005) West Nile virus inhibits the signal transduction pathway of alpha interferon. J Virol 79(3):1343–1350
Halpin K, Young PL, Field H, Mackenzie JS (1999) Newly discovered viruses of flying foxes. Vet Microbiol 68(1–2):83–87
Hughes AL (1994) The evolution of functionally novel proteins after gene duplication. Proc Biol Sci 256(1346):119–124
Hughes AL (1995) The evolution of the type I interferon gene family in mammals. J Mol Evol 41(5):539–548
Hughes AL (2000) Adaptive evolution of genes and genomes. Oxford University Press, New York
Hughes AL, Hughes MK (1993) Adaptive evolution in the rat olfactory receptor gene family. J Mol Evol 36(3):249–254
Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol 7(2):131–137
Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Délicat A, Paweska JT, Gonzalez JP, Swanepoel R (2005) Fruit bats as reservoirs of Ebola virus. Nature 438(7068):575–576
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF (2005) Bats are natural reservoirs of SARS-like coronaviruses. Science 310(5748):676–679
O’Shea JJ, Gadina M, Schreiber RD (2002) Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109(Suppl):S121–S131
Ohno S, Wolf U, Atkin NB (1968) Evolution from fish to mammals by gene duplication. Hereditas 59(1):169–187
Pestka S, Langer JA, Zoon KC, Samuel CE (1987) Interferons and their actions. Annu Rev Biochem 56:727–777
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32
Reid SP, Leung LW, Hartman AL, Martinez O, Shae ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF (2006) J Virol 80(11):5156–5167
Samuel CE (2001) Antiviral actions of interferons. Clin Microbiol Rev 14(4):778–809
Sawyer S (1989) Statistical tests for detecting gene conversion. Mol Biol Evol 6(5):526–538
Schultz U, Kaspers B, Staehili P (2004) The interferon system of non-mammalian vertebrates. Dev Comp Immunol 28(5):499–508
Shaw GD, Boll W, Taira H, Mantei N, Lengyel P, Weissmann C (1983) Structure and expression of cloned murine IFN-alpha genes. Nucleic Acids Res 11(3):555–573
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad USA 101(30):11030–11035
Tamura K, Dudley J, Nei M, Kumar S (2007) Mega4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24(8):1596–1599
Taniguchi T, Takaoka A (2001) A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2(5):378–386
Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Woelk CH, Frost SD, Richman DD, Higley PE, Kosakovsky Pond SL (2007) Evolution of the interferon alpha gene family in eutherian mammals. Gene 397(1–2):38–50
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15(5):568–573
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24(8):1586–1591
Yeow WS, Lawson CM, Beilharz MW (1998) Antiviral activities of individual murine IFN-alpha subtypes in vivo: intramuscular injection of IFN expression constructs reduces cytomegalovirus replication. J Immunol 160(6):2932–2939
Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, Ksiazek T (2001) Nipah virus infection in bats (order Chiroptera) in Peninsular Malaysia. Emerg Infect Dis 7(3):439–441
Acknowledgments
We thank an anonymous reviewer for valuable comments. This work was supported by PhD Program Scholarship Fund of ECNU (2010044) to Jie Cui; and a start-up fund (77102175) from ECNU to Guimei He.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material. The online version of this article contains supplementary material, which is available to authorized users)
Rights and permissions
About this article
Cite this article
He, G., He, B., Racey, P.A. et al. Positive Selection of the Bat Interferon Alpha Gene Family. Biochem Genet 48, 840–846 (2010). https://doi.org/10.1007/s10528-010-9365-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-010-9365-9