
Abstract
Physical map of chickpea was developed for the reference chickpea genotype (ICC 4958) using bacterial artificial chromosome (BAC) libraries targeting 71,094 clones (~12× coverage). High information content fingerprinting (HICF) of these clones gave high-quality fingerprinting data for 67,483 clones, and 1,174 contigs comprising 46,112 clones and 3,256 singletons were defined. In brief, 574 Mb genome size was assembled in 1,174 contigs with an average of 0.49 Mb per contig and 3,256 singletons represent 407 Mb genome. The physical map was linked with two genetic maps with the help of 245 BAC-end sequence (BES)-derived simple sequence repeat (SSR) markers. This allowed locating some of the BACs in the vicinity of some important quantitative trait loci (QTLs) for drought tolerance and reistance to Fusarium wilt and Ascochyta blight. In addition, fingerprinted contig (FPC) assembly was also integrated with the draft genome sequence of chickpea. As a result, ~965 BACs including 163 minimum tilling path (MTP) clones could be mapped on eight pseudo-molecules of chickpea forming 491 hypothetical contigs representing 54,013,992 bp (~54 Mb) of the draft genome. Comprehensive analysis of markers in abiotic and biotic stress tolerance QTL regions led to identification of 654, 306 and 23 genes in drought tolerance “QTL-hotspot” region, Ascochyta blight resistance QTL region and Fusarium wilt resistance QTL region, respectively. Integrated physical, genetic and genome map should provide a foundation for cloning and isolation of QTLs/genes for molecular dissection of traits as well as markers for molecular breeding for chickpea improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chickpea is a self-pollinated diploid (2n = 2x = 16) annual grain legume with a genome size of approximately 740 Mbp (Arumuganathan and Earle 1991). It is cultivated mostly on residual soil moisture in semi-arid regions of South Asia and sub-Saharan Africa. India is the largest producer of chickpea contributing about 60 % of total world’s production. Other important chickpea producing countries are Pakistan, Turkey, Mexico, USA, Canada and Australia (http://www.cgiar.org/our-research/crop-factsheets/chickpea/). The seeds of chickpea are rich in protein (24.6 %) and carbohydrate (64.6 %) and a good source of minerals and fibers and a source of food grain with low cholesterol levels (Abu-Salem and Abou 2011). There are two distinct chickpea types, Desi and Kabuli, different in their morphology. Desi-type chickpeas possess purple flower and small, dark and angular seeds and are largely consumed in Indian subcontinent and Pakistan, while Kabuli chickpeas have white flowers and large, cream-coloured seeds and are preferred in the Mediterranean basin and Central Asia where they are mainly consumed as a whole seed. Despite the increase in area and production during the last decade, there has not been a significant increase in the productivity of chickpea (Varshney et al. 2010). Considerable breeding efforts have been made across the globe to overcome the abiotic (drought, salinity, heat) and biotic (Helicoverpa, Fusarium wilt, Ascochyta blight) production constraints in chickpea. The genetic gains obtained through the conventional breeding efforts are not on par with the growing demands of the crop. Therefore, molecular breeding is now becoming one of the integral components of chickpea breeding programs (Chamarthi et al. 2011; Varshney et al. 2013a).
Recent advances in chickpea genomics have made it possible to not only develop large-scale molecular markers, genetic maps, transcriptomic resources for conducting large-scale and high-throughput marker genotyping but also sequence the genome of this important crop (Varshney et al. 2013b; Jain et al. 2013). For instance, >3,000 simple sequence repeat (SSR) markers (Nayak et al. 2010; Thudi et al. 2011; Choudhary et al. 2012), 15,360 Diversity Arrays Technology (DArT) loci (Thudi et al. 2011) and >3,000 single nucleotide polymorphism (SNP) markers (Hiremath et al. 2012; Gaur et al. 2012) have been developed. Several genetic maps have been constructed (Nayak et al. 2010; Millán et al. 2010; Gujaria et al. 2011; Thudi et al. 2011; Choudhary et al. 2012; Hiremath et al. 2012; Gaur et al. 2012), and hundreds of quantitative trait loci (QTLs) for drought tolerance (Varshney et al. 2014), salinity (Vadez et al. 2012), Ascochyta blight (Kottapalli et al. 2009; Anbessa et al. 2009), Botrytis grey mould (Anuradha et al. 2011) and Fusarium wilt (Tekeoglu et al. 2002; Udupa and Baum 2003) have been mapped. Further, large-scale expressed sequence tags (ESTs; Varshney et al. 2009) were generated and transcriptome assembly-constructed (Garg et al. 2011; Hiremath et al. 2012; Kudapa et al. 2014). In addition, several large-insert bacterial artificial chromosome (BAC) and binary bacterial artificial chromosome (BIBAC)-based libraries were also constructed for chickpea (Lichtenzveig et al. 2005; Zhang et al. 2010). The available genomic resources for chickpea molecular breeding community have been recently reviewed extensively by Upadhyaya et al. (2011) and Varshney et al. (2012).
In the context of development of physical maps, a BAC/BIBAC-based physical map was developed (Zhang et al. 2010). On this physical map, three large contigs closely linked to QTLs contributing to Ascochyta blight resistance and flowering time in chickpea were identified (Zhang et al. 2010). However, a comprehensive genome-wide physical map, and its integration with genetic maps possessing QTLs for important targeted traits and draft genome of chickpea, is the need of the hour for facilitating cloning of candidate genes and enhancing molecular breeding programs in this important crop.
In this study, we have constructed a genome-wide physical map employing large-insert BAC libraries and integrated it with the genetic maps and chickpea reference genome sequence for the identification of BAC clones covering important genomic regions for drought tolerance, Ascochyta blight and Fusarium wilt resistance. A comprehensive analysis of the targeted QTL regions in the integrated genetic, physical and genome maps has provided candidate genes that may be used for functional analysis as well as molecular breeding for drought tolerance and resistance to Ascochyta blight and Fusarium wilt.
Materials and methods
Plant material
ICC 4958, a drought tolerant Desi chickpea genotype, was used for the construction of BAC libraries. DNA was isolated from etiolated young seedling as per Cuc et al. (2008).
BAC libraries
Two BAC libraries were constructed from high molecular weight (HMW) genomic DNA processed at Amplicon Express, Pullman, Washington, using the method of Tao et al. (2002). Purified large DNA fragments were ligated with pCCBAC1 (Epicentre, Omaha, NE, USA). Ligations were transformed into DH10B Escherichia coli cells (Invitrogen, Grand Island, NY, USA) and plated on Luria-Bertani (LB) agar with appropriate chloramphenicol, X-gal and IPTG concentrations. Clones were robotically picked with a Genomic Solutions G3 into 384-well plates containing LB freezing media. Plates were incubated for 18 h, replicated and then frozen at −80 °C. The replicated copy was used as a source plate for physical mapping.
High information content fingerprinting
We used the four-colour high information content fingerprinting (HICF) SNaPshot method of Luo et al. (2003) with modification suited for ABI Genetic Analyzer 3730 platform (Gu et al. 2009). For each 384-well plate from the library, four 96-well blocks were inoculated using a 96-pin replicator. To control for plate orientation and fingerprinting, quality wells E7 and H12 were replaced in each 96-well block with a control BAC clone. The cultures were grown for 20 h at 37 °C and 400 rpm on an orbital shaker. The BAC DNA was purified using the Qiagen R.E.A.L. 96 Prep Kit (Qiagen, Valencia, CA, USA). Each BAC was simultaneously digested with four 6-bp recognizing restriction endonucleases generating 3′ recessed ends. Each of the four recessed 3′ ends was labeled with a different fluorescent dye using the SNaPshot kit (Applied Biosystems, Foster City, CA, USA). Restriction fragments were sized with a capillary DNA analyzer ABI 3730XL (Applied Biosystems, Foster City, CA, USA) using an internal GeneScan 1200 LIZ size standard. Fragment size calling was performed with the GeneMapper v. 3.7 (Applied Biosystems, Foster City, CA, USA).
Fingerprinting data editing
Outputs of size-calling files from GeneMapper software were automatically edited with the FP Miner program (Gu et al. 2009). This software package was used to distinguish peaks corresponding to restriction fragments from peaks generated by a background noise in the profile of each BAC fingerprint and to remove vector restriction fragments from the profiles. The program also removed substandard profiles that could negatively affect contigs assembly. The files generated by FP Miner were used in the fingerprinted contig (FPC) assembly.
Physical map contig assembly
Contigs were assembled from fragments within a size range of 70–1,000 bp using FPC software (version 9.3, http://www.agcol.arizona.edu/software/fpc/), using the following assembly strategy. We set tolerance at 6 (0.6 bp) and an initial cut-off of 1E − 60 (1 × 10−60). We followed the initial assembly with several rounds of DQer, until no contig containing 15 % or more questionable (Q) clones. This was followed by several rounds of end-to-end merging and single-to-end merging at progressively lower cut-off stringencies. The “best of” function was set to 100 builds.
FPC displays the length of each BAC clone and each BAC contig in consensus band (CB) units. To convert CB units to kilobytes, we estimated the lengths of inserts in 100 BAC clones in kb by pulsed field gel electrophoresis (PFGE) and divided the total length by the number of restriction fragments in the fingerprints of the clones. The conversion factor for the BAC clones was 1.92 kb/CB unit.
Anchoring of physical map with genetic maps
For anchoring the physical map with the genetic map, a set of 337 BAC clones corresponding to 337 polymorphic BAC-end sequence-derived SSR (BES-SSR) markers in inter-specific genetic map (ICC 4958 × PI 489777; Thudi et al. 2011) and two intra-specific genetic maps (ICC 4958 × ICC 1882 and ICC 283 × ICC 8261) were subjected to fingerprinting in the same way as mentioned above. The fingerprinted clones were tried for their assembly with the FPC contig physical map for anchoring genetic map with the physical map.
Alignment of physical map with draft genome of chickpea
A set of 4,290 BAC clones forming the minimum tilling path (MTP) of FPC contig were picked, and plasmid DNA was isolated as described by Huo et al. (2008). BAC clones were sequenced with BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA) using the pIndigoBAC-5 reverse primer (5′-CTCGTATGTTGTGTGGAATTGTGAGC-3′). Amplifications were carried out in 96-well plate (10 μl reaction volumes containing 1 μl of BigDye, 1.75 μl sequencing reaction buffer, 2 mM MgCl2, 5 % DMSO, 0.5 μM primer and 200–500 ng of BAC DNA) or 384-well plate (5 μl reaction volume). PCR was performed with 49 cycles of 96 °C for 10 s, 50 °C for 10 s and 60 °C for 4 min. The PCR product was purified and then analyzed on the 3730XL DNA analyzer (Applied Biosystems, Foster City, CA, USA). Base calling of ABI trace file was performed with PHRED (Ewing and Green 1998; Ewing et al. 1998), and sequences with quality scores of less than 20 were trimmed. Vector sequences were removed using Cross_match. BES less than 100 bp was also filtered.
Similarly, another set of 46,270 BAC-end sequences for CAH1 library that were used to develop novel SSR markers and development of high-density genetic map in chickpea were also used for in silico mapping onto draft chickpea genome during the present study. The details of construction of BAC library, BAC-end sequences is available elsewhere (Thudi et al. 2011).
In addition, efforts have also been made to map onto draft genome sequence the genetically mapped 1,328 marker loci including novel 625 Chickpea KASPar Assay Markers (CKAMs) and 314 tentative orthologous genes (TOGs)-SNPs from Hiremath et al. (2012) for integrating physical, sequence and genetic maps. For repeat analysis, a repeat database for chickpea genome was first built from the Kabuli draft genome already available with us (Varshney et al. 2013b), followed by search for different types of repeat sequences from the 53,316 BESs using RepeatMasker v 3.2.7 (Tarailo-Graovac and Chen 2009). The unique sequences were isolated from repetitive ones using NCBI BLAST+ ver 2.2.25 with a cut-off E value of less than 10−50. All the 53,316 BESs were used as Basic Local Alignment Search Tool (BLAST) query against chickpea repeat database.
The clean sequences after repetitive sequence analysis were subjected to mapping onto draft genome of Kabuli chickpea using NCBI BLAST+. From all of the BLAST alignments, clean BESs were extracted according to the following criteria (Katagiri et al. 2004; Asamizu et al. 2012): (i) sequence identity >90 % and alignment coverage >50 %, (ii) mapped positions of each pair of ends >100 bp and <200 kb apart in the same chromosome, (iii) direction of each paired end is correct, (iv) BLASTN E < 10−100, (v) a minimum of one hit for one of the paired ends and (vi) no redundant chromosomal locations.
Identification of genes in important QTL regions
In order to identify candidate genes present in the QTL regions for some important traits such as drought tolerance (“QTL-hotspot”, Varshney et al. 2014), Ascochyta blight resistance (Udupa and Baum 2003) and Fusarium wilt resistance (Sabbavarapu et al. 2013), the markers present in these QTL regions were subjected to BLAST against chickpea genome assembly (Varshney et al. 2013b) and the corresponding UniProt IDs were retrieved. For functional categorization of the genes, the UniProt IDs of the genes were mapped onto UniProt KB database (http://www.uniprot.org/).
Results
New BAC libraries
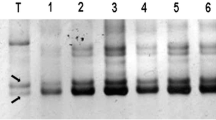
Two BAC libraries were constructed from genomic DNA of chickpea Desi cultivar “ICC 4958” using HindIII and EcoRI restriction enzymes and were designated as CAH0000 and CAE0000, respectively. From each of these two libraries, 48,384 clones were picked up, and in total, 96,768 clones were obtained. For quality control, four plates were randomly selected from each library, and 24 clones from various quadrants of each plate were picked up. In brief, 96 random clones were selected from both CAE0000 and CAH0000 BAC libraries for quality control analysis. These analyses revealed average insert sizes of 120 kb in CAH0000 library and 124 kb in CAE0000 library and approximately eight-fold coverage of the chickpea haploid genome in each of these two libraries (Fig. 1a, b). It was also observed that ~3 to 4 % of clones in both libraries contained no inserts of chickpea DNA.

Insert size estimation of chickpea BAC clones by pulsed field gel electrophoresis (PFGE). A set of 28 clones can be visualized from a CAH0000 library (constructed using HindIII) and b CAE0000 library (constructed using EcoRI). PFG marker can be seen in two lanes on either side of the 28 clones from each library. The insert size of BAC library is ~120 and ~124 kb for CAH0000 and CAE0000 libraries, respectively
In addition, another BAC library (CAH1) constructed from the chickpea cultivar ICC 4958 using HindIII restriction enzyme (Thudi et al. 2011) was also used in the present study. From this library, a set of 1,110 BACs containing resistance gene homolog (RGH) and BES-SSRs were used for developing the physical map.
BAC fingerprinting
A set of 71,094 clones (~12× coverage) including 35,040 clones from CAH0000, 34,944 clones from CAE0000 library, 337 BES-SSR-containing clones and 773 disease resistance gene-containing clones from CAH1 library (Thudi et al. 2011) were targeted for fingerprinting (Table 1). Fingerprints were obtained for 68,248 clones (95.99 %). After removing cross-contaminated clones, clones with imperfect fingerprints and clones possessing small inserts, 49,368 clone fingerprints were found to be suitable and were used for contig assembly (Table 1). Number of bands from each clone varied from 34 to 2,268 (Table 2).
Physical map assembly
FPC version 9.3 was used for assembling a physical map. A total of 1,174 contigs could be assembled from 49,368 clones with usable fingerprinting data with the procedure described in the “Materials and methods” section (Table 2). The 1,174 contigs contained 46,112 clones, of all of which, a set of 4,290 clones defined the MTP of physical map assembly, while 3,256 clones remained as singletons. The range of clones in each contig varied from 2 to 3,007 with an average of 39.27 clones/contig (Table 2, Fig. 2; http://phymap.ucdavis.edu/chickpea/).

Distribution of number of clones in 1,174 contigs in chickpea FPC assembly. The range of clones in each contig varied from 2 to 3,007 with an average of 39.27 clones/contig
An analysis of Q clones in our data set indicated that 443 contigs out of total 1,174 contigs (37.73 %) contained a total of 2,093 Q clones (2,093 / 49,368 = 4.23 %) with a range of 1 to 89 Q clones/contig (Table 2). The low number (4.23 %) of Q clones in our assembly indicated the good quality and proper cut-off and tolerance values used for the assembly.
Anchoring physical map with genetic maps
In order to anchor the contig map with the genetic map, a total of 337 polymorphic BES-SSRs were used for mapping on three mapping populations (one inter- and two intra-specific crosses). However, only 259 BES-SSR markers were integrated on genetic maps based on three populations, namely, ICC 4958 × PI 489777 (inter-specific mapping population) and ICC 4958 × ICC 1882 and ICC 283 × ICC 8261 (intra-specific populations) (Supplementary Table 1). A consensus map was also developed based on genetic maps for intra-specific mapping populations (http://cmap.icrisat.ac.in/cmap/sm/cp/varshney/).
BAC clones of all 337 polymorphic BES-SSRs were subjected for fingerprinting as mentioned above, and 319 were assembled into FPC assembly (Table 1). However, only 259 mapped markers on above-mentioned genetic maps could be aligned onto the physical map. These data were used for anchoring genetic maps with the physical map (Supplementary Table 1). A summary of distribution of these 259 mapped markers on different linkage groups (LGs) is tabulated in Table 3. The details of contigs hit by these BES-SSRs of chickpea FPC assembly have been given in Supplementary Table 1 and in Fig. 3.

Anchoring physical map with two chickpea genetic maps. The number of marker hits on BAC contigs varied from 1 to 5; unique hits were obtained for 131 markers
In addition to above, 619 genetically mapped marker loci including CKAMs and TOGs that were mapped onto chickpea draft genome sequence/scaffolds were found very close/linked to all 163 MTP clones of physical map mapped onto eight pseudo-molecules of draft genome sequence. This has also helped to anchor a physical map with the second-generation genetic map developed based on cost-effective SNP marker assays (Hiremath et al. 2012).
QTLs assigned to integrated genetic and physical map
A number of QTLs were identified for abiotic and biotic stresses earlier; in addition, we also identified QTLs for abiotic stress (drought tolerance, salinity) and biotic stress (like Fusarium wilt, Ascochyta blight and Botrytis grey mould) from published studies, and their corresponding location on the inter-specific map based on ICC 4958 × PI 489777 (Thudi et al. 2011) was determined (Supplementary Table 1).
While analyzing the physical map in details, several BAC clones in proximity to "QTL-hotspot" were identified on CaLG04, and its traits were identified. The “QTL-hotspot” possesses QTLs for root traits (root length density, root volume, root surface area, root length, root dry weight, shoot dry weight, leaf dry weight), phenological traits (days to flowering and days to maturity), yield, harvest index, biomass, 100-seed weight, seeds per pod and carbon isotope discrimination under both rain-fed and irrigated conditions (Varshney et al. 2014). A BAC contig (ctg198) has been found very close (2.1 cM) to the associated/linked marker TAA170 on the intra-specific genetic map of ICC 4958 × ICC 1882, and BAC contig (ctg1769) was found very close to ICCM0249 markers on the linkage group CaLG04 of an inter-specific genetic map of ICC 4958 × PI 489777 (Fig. 4).

A snapshot of anchoring BAC clones to the “QTL-hotspot” region harbouring QTLs for several drought tolerance-related traits in chickpea. Two BAC contigs namely ctg198 and ctg1769 were assigned to “QTL-hotspot” region as SSR markers namely CaM0232 and CaM1328 derived from end sequences of BACs from the above mentioned contigs were mapped in the same region on inter-specific (Thudi et al. 2011) and intra-specific (Varshney et al. 2014) genetic maps, respectively
A BAC contig closer to the Ascochyta blight resistance QTLs reported by Aryamanesh et al. (2010) and Kottapalli et al. (2009) was identified. Similarly, several other BAC contigs were found to be closer or in the same QTL regions. For instance, ctg1390 (260 kb) was found closer to ar1 and ar2a on LG02, QTLs for Ascochyta blight resistance, and similarly, ctg298 (4,290 kb) was found close to ar2b QTL on LG04 reported by Udupa and Baum (2003) (Fig. 5). One contig, ctg248, was identified in close proximity to the Fusarium oxysporum f. sp. ciceris race 0 locus reported by Cobos et al. (2007). Fourteen contigs were also found in close proximity to the beta-carotene-related QTLs (Supplementary Table 1).

A snapshot showing coverage of Ascochyta blight resistance QTL regions with BAC clones in chickpea. The BAC ctg1390 on LG2 of inter-specific genetic map developed by Thudi et al. (2011) based on RIL population ICC 4958 × PI 489777 is closer to the SSR marker GA16 that flanks the ar2a QTL identified for Ascochyta blight resistance by Udupa and Baum (2003). Similarly, the BAC contig 198 on LG04 of inter-specific genetic map developed by Thudi et al. (2011) is closer to SSR marker TA130 that flanks ar2b QTL identified for Ascochyta blight resistance by Udupa and Baum (2003)
Anchoring physical map with the draft genome of Kabuli chickpea
For anchoring a contig physical map with the draft genome sequence of chickpea (sequence map; Varshney et al. 2013b), a set of 53,316 BESs (37.8 Mb including redundancy) from 28,147 clones representing ~5.12 % chickpea genome were used (Table 4). BESs ranged from 64 to 1,113 bp with an average of 708 bp. Among 53,316 BESs, 50,338 BES (94 %) were found to be mate pairs from 25,169 clones and 2,978 BESs (6 %) were single ends of BAC clones (Table 4). The repeat analyses of all the BESs showed ~70 % of sequences which constitute different kinds of repeats with abundance (~60 %) of LTR elements (Supplementary Table 2). Further analysis of BESs provided 16,086 unique (non-repeat) BES, while 37,230 BESs were found to contain repeat BESs and were therefore excluded from further mapping (Fig. 6). The unique BESs (16,086; 30 %) were tried for their mapping onto the chickpea genome sequence covering eight pseudo-molecules (Ca1 to Ca8) and un-anchored scaffolds (Ca0). A total of 965 BAC clones could be successfully mapped onto a draft genome of Kabuli chickpea (Table 5; Supplementary Fig. 1a–h). The mapped BAC clone size distribution on the basis of results revealed 428 out of 965 (44.35 %) BACs ranged from 90 to 110 kb with an average size of 95 kb. The number of BAC clones mapped on individual pseudo-molecule ranged from 64 on Ca8 to 154 on Ca4 with an average of ~110 BAC clones/pseudo-molecule (Table 5). A set of 85 BACs were mapped on scaffolds not assigned to any pseudo-molecule (Ca0). The number of BAC clones mapped on chickpea genome sequence consists of 491 contigs covering a total of ~55 Mb physical length (Table 5). The number of contigs on each pseudo-molecule ranged from 31 on Ca8 to 93 on Ca4 with an average of ~62 contigs/pseudo-molecule (Table 5). All these contigs represent only a small proportion (~15 %) of the genome represented by eight pseudo-molecules.

Summary of steps involved in BES mapping analysis for integrating chickpea FPC contig assembly with the high-density genetic map and the reference chickpea genome sequence. A set of 53,316 BESs from 28,147 clones were used for anchoring contig physical map with the draft genome sequence of chickpea. BLASTN analysis of 53,316 BESs provided 16,086 unique (non-repeat) BES. The unique BESs (16,086) were tried for their mapping onto the chickpea genome sequence covering eight pseudo-molecules (Ca1 to Ca8) and un-anchored scaffolds (Ca0). As a result, a total of 965 BAC clones were successfully mapped onto draft genome of Kabuli chickpea, while 13,868 BES had multiple hits, and 1,252 had either low similarity or no hits
Similarly, we also tried to map sequences of 1,328 genetically mapped loci from Hiremath et al. (2012) onto the daft genome sequence of chickpea. As a result, 619 CKAMs and TOGs were mapped onto the chickpea draft genome sequence/scaffolds and were found to be very close/linked to MTP clones of a physical map mapped onto draft genome sequence. The number of mapped marker loci on the draft genome sequence ranged from 43 (on Ca8) to 113 (on Ca4) with an average of ~77 loci/pseudo-molecule (Supplementary Fig. 1a–h). This helped us to anchor a physical map with the draft genome sequence of chickpea and genetic maps.
Mining candidate genes in select stress responsive QTL regions
After identification of BAC contigs/clones in the region or proximity QTLs for drought tolerance (“QTL-hotspot”), Ascochyta blight and Fusarium wilt resistance, an effort was made to mine candidate genes in the genomic regions after aligning molecular markers present in/associated with the QTL regions on genome map/sequence assembly. For instance, in the case of “QTL-hotspot” region, analysis of seven SSR markers (ICCM0249, NCPGR127, TAA170, NCPGR21, TR11, STMS 11 and GA24) on CaLG04 with a genome sequence identified a region of ca. 7 Mb (starting at 9.1 Mb and ending at 16.1 Mb) on Ca4 chromosome. Genome annotation of this region has identified a total of 654 genes (Fig. 5; Supplementary Table 3). In the case of Ascochyta blight resistance, analysis of two SSR markers (GA16 and CaM1135) present in QTL region/genes ara1 and ara2a (Thudi et al. 2011) identified a 3 Mb region (starting at 31 Mb and ending at 34 Mb) on Ca2 that contained a total of 306 genes (Supplementary Table 4). However, the markers TA130 and CaM0232 on LG4 (Thudi et al. 2011; Fig. 5) corresponding to ara2b QTL/gene could not be mapped; hence, this QTL region was not analyzed in details. For Fusarium wilt resistance QTL region, analysis of two markers (CaM1125 and TR44; Sabbavarapu et al. 2013) on genome sequence provided a relatively smaller region (0.6 Mb, between 29 and 29.6 Mb), comprising 23 genes (Supplementary Table 5).
All 983 genes (654 in “QTL-hotspot” region, 306 in Ascochyta blight resistance QTL region and 23 in Fusarium wilt resistance QTL region) were functionally categorized based on Gene Ontology (GO) descriptions (UniProt database, 151 UniProt-GO), and all could be assigned to at least one GO term (Table 6). The genes in all three QTL regions were further assigned to three functional categories: (i) molecular function, (ii) cellular component and (iii) biological processes.
In the case of “QTL-hotspot” region, out of 654 genes, 362 genes were assigned to “molecular function” category, 252 genes to “cellular component” category and 370 genes to “biological process” category, respectively. It is important to note here that the sum of genes assigned to different functional categories (984) is higher than the total number of genes (654), as a given gene may fall in more than one category. In the molecular function category, the highest number of genes fell into catalytic activity (239) followed by binding (219). Under cellular component category, the highest number of genes fell into cell part (190) followed by organelle (160). Similarly, in the biological processes category, a maximum number of genes fell into metabolic process (296) followed by cellular process (239).
For Ascochyta blight QTL region, 157 genes were assigned to molecular function, 112 genes to cellular component and 162 genes to biological processes. The highest number of genes under molecular function category fell into catalytic activity (95). In the cellular component category, a maximum number of genes (88) belonged to cell part class. Similarly, under biological processes category, the highest number of genes fell to metabolic process class that contained the highest number of genes (118).
In the case of Fusarium wilt resistance QTL region, 14 genes were assigned to molecular function category, 9 genes to cellular component category and 14 genes to biological processes, respectively. Among these categories, binding class with 11 genes, cell part with 9 genes and cellular process with 12 genes were found in abundance under molecular function, cellular component and biological process categories, respectively.
Discussion
Chickpea is one of the important diploid legume crops (with moderate genome size of 738 Mb) for the poor, largely vegetarian people living in semi-arid tropics (SAT) and other climate vulnerable regions in the world. The availability of physical maps in crop plants serves as an indispensable tool and have been used for a variety of purposes including (i) QTL fine mapping and positional (map-based) cloning of QTLs/genes (Bakker et al. 2003), (ii) anchoring chromosomes using fluorescence in situ hybridization (FISH) (Islam-Faridi et al. 2002), (iii) repeat classification (Cardle et al. 2000), (iv) draft genome sequence assembly (Sasaki et al. 2005), (v) marker development experiments (van der Vossen et al. 2000) and (vi) analysis of structural variation in the genome (Kidd et al. 2008). In the recent past, significant progress and tremendous advancements have been made in the area of sequencing/resequencing crop genomes using next-generation sequencing (NGS) technologies (Hillier et al. 2008; Wheeler et al. 2008; Thudi et al. 2012). However, despite the advancements in NGS technologies, the need and value for development of high-quality physical maps still remains high (Lewin et al. 2009). For instance, the sequencing/assembly of crops possessing complex genomes with large fractions of repetitive genome will not be easily addressed by NGS alone but will be facilitated by physical maps which would provide anchor points to link sequence contigs and bridge gaps due to large repeat regions. One of the effective ways for providing these anchor points is by construction of a whole genome physical map using bacterial artificial chromosome (BAC) clones (Shizuya et al. 1992; Rounsley et al. 2009) and BAC-end sequencing (Nelson et al. 2005). The development of physical maps using BAC clones has been proven effective for the construction of genome-wide physical maps, since generation and storage of these clones is somewhat relatively easy (Gregory et al. 1997; Marra et al. 1997; Klein et al. 2000; Wu et al. 2004). Therefore, BAC-based physical maps combined with BAC-end sequencing have formed the basis of several whole genome sequencing projects (Sasaki et al. 2005; Wei et al. 2007). Keeping the importance of physical maps in view, a BAC-based physical map has been developed for chickpea in the present study.
Physical map assembly and genome coverage/representation of chickpea genome
In the present study, we reported the construction of a high-quality, high-coverage BAC-based physical map of chickpea genome using genomic DNA of cultivar ICC 4958. The physical map has a total of 49,368 BAC clones assembled into 1,174 contigs. The number of clones in each contig varied from 2 to 3,007 with an average of ~39 clones/contig spanning from >200 kb to 4.1 Mb, with an average physical length of ~489 kb (574 / 117 = 0.4889 Mb). These contigs collectively span 615 Mb, smaller than the expected 738 Mb estimated genome size of chickpea genome by 17 %.
The genome coverage (eight times) of chickpea genome (46,112 clones) assembled into chickpea physical map assembly (Table 2) was sufficient. Comparative genome coverage (10× coverage) has been found earlier to be sufficient for developing a physical map of the chickpea genome (Zhang et al. 2010). Similarly, the number of clones used for fingerprinting (70,321) has been also found sufficient for generating a quality physical map, since less number of clones (67,584) were fingerprinted in a previous study of chickpea physical map development (Zhang et al. 2010).
The chickpea physical map was developed using two BAC libraries based on patterns of fragments generated by digestion of restriction enzymes (BamHI, EcoRI, XbaI, XhoI and HaeIII). The genome size estimated by K-mer analysis revealed a genome size of 738 Mb. However, genome size of only 615 Mb could be assembled into contigs, while 300 Mb remained in the form of singletons. The estimation of coverage of physical map was based on contigs only, and if singletons were also included, then the genome coverage of chickpea physical map might be overestimated (615 + 300 = 915 Mb). The discrepancy in genome size estimation is not unusual as in earlier studies; also, overestimation of genome size has been reported such as in Populus (about 20 % larger; Kelleher et al. 2007) and soybean (26.3 % larger; Wu et al. 2004). However, in the case of papaya, the genome size estimated based on the physical map was close to the actual size (Yu et al. 2009). The smaller genome size obtained in the present study may be attributed to the following: (i) false end-merges of clones and formation of large contigs; (ii) more overlaps between contigs; (iii) 3,256 or 6.5 % of clones remained as singletons, and 4.4 % (2,137) of the clones were located on small contigs (containing fewer than ten clones); (iv) average insert size estimation was not perfect based on representative samples of BAC libraries; (v) genome size of chickpea not perfectly estimated earlier; and (vi) the use of only two restriction enzymes for BAC library construction, since the use of more number of RE in BAC library construction has been found to increase the actual coverage of map (Zhang et al. 2010). The existence of more overlap than the expected between the contigs will reduce the overall size of contigs and, hence, the physical map assembly. Discrepancies in genome size of chickpea was also reported in an earlier study of the development of physical map in chickpea; however, they reported more genome size than the expected 738 Mb (Zhang et al. 2010). Despite the genome size estimation discrepancy, the quality of the chickpea physical map developed in the present study was found to be sufficiently high for use in various applications of genomics research.
Integrating physical map with the genetic/QTL maps
A genetic linkage map is constructed by placing genetic loci on chromosomes based on recombination frequencies, while a physical map is constructed based on overlapping restriction patterns of large insert BAC clones. Therefore, integration of these two different types of maps may reveal recombination hotspots as well as regions suppressed for recombination. In addition, this may allow an estimation of physical distances between genetic markers, thereby providing a framework for assembling the whole genome shotgun sequences.
Keeping in view the importance of integrating physical maps with genetic maps, we tried to anchor our physical map assembly with the genetic map by fingerprinting of 337 BAC clones and assembly of 319 of these clones into physical map assembly. The SSR markers derived from BAC-end sequencing of these clones had been found to be polymorphic and were mapped in the three bi-parental mapping populations derived from crosses ICC 4958 × PI 489777, ICC 4958 × ICC 1882 and ICC 283 × ICC 8261. Therefore, integrating these BAC clones into the physical map assembly helped to anchor our physical map with these three genetic maps (Supplementary Table 1).
The integration of physical map with genetic maps has been reported earlier in different model plant species including Arabidopsis (Meinke et al. 2009), rice (Chen et al. 2002) and fruit trees including peach (Zhebentyayeva et al. 2008), papaya (Yu et al. 2009) and apple (Han et al. 2011). The anchoring of physical map with the genetic map helped us to identify the BAC clones within or close to some of the QTLs already reported for important traits in chickpea (Supplementary Table 1). Therefore, the chickpea physical map may greatly help in cloning and fine mapping of some of these important genes in the future, thereby facilitating molecular breeding in this important crop.
Further, additional efforts are necessary to integrate and correlate the physical contig map with the other available genetic maps of chickpea for locating all mapped genes and QTLs to physical map contigs. These efforts have been further accelerated with the recent availability of the whole genome sequences of chickpea from NIPGR, New Delhi, India (Jain et al. 2013), and by the International Chickpea Genome Sequencing Consortium (Varshney et al. 2013b).
Integrated physical and genetic map is expected to act as an important resource for expediting the mapping and cloning of target genes controlling complex quantitative traits like drought and yield in chickpea. The availability of gene information will facilitate molecular breeding for improving important targeted traits in this important legume crop.
Integrating physical map with the Kabuli chickpea genome map
Genome sequencing of all the model legumes including Lotus (Sato et al. 2008), soybean (Schmutz et al. 2010) and Medicago (Young et al. 2011) has been completed. Following model genomes, several other legume genomes have been sequenced including pigeon pea (Varshney et al. 2012), common bean (Scott Jackson, personal communications) and, recently, chickpea (Varshney et al. 2013b), and efforts are being made to sequence peanut, lentil and cowpea. This revolution in whole genome sequencing of crop genomes has made the integration of physical maps with the draft genome sequence possible. Chromosome-anchored physical maps may serve as an important tool for (i) improving the draft genome sequence by filling sequence gaps; (ii) resolving assembly errors caused due to repetitive sequences, large gene families and segmental duplications; (iii) providing anchor pints; (iv) ordering of sequence contigs/scaffolds; (v) map-based cloning; and (vi) cloning sequences that are too large or repetitive for PCR-based cloning (Ha et al. 2012).
Mapping of BESs from one variety onto the reference genome sequence of a different variety may help to identify putative chromosomal structural rearrangements between the two varieties. This has been done successfully for the identification of structural rearrangements between Glycine soja and Glycine max by aligning BESs from G. soja against the G. max reference sequence (Ha et al. 2012). Thus, physical maps will also help to investigate a structural evolution that has occurred between two genomes and to allow researchers to effectively shuttle between the genomes to capture useful information for crop improvement and basic genetics research (Ha et al. 2012).
The availability of genome sequence of CDC Frontier, a Canadian Kabuli chickpea variety (Varshney et al. 2013b), has facilitated the integration of BESs from the physical map including BES of MTP clones form the chickpea cultivar ICC 4958 with the daft sequence of chickpea. A set of only ~965 BACs including 163 MTP clones could be mapped onto the draft genome of chickpea. These BACs formed 491 hypothetical contigs representing 54,013,992 bp (~54 Mb) of reference genome. The mapping of 163 MTP clones on eight chickpea pseudo-molecule ranged from 11 (on Ca8) to 29 (Ca4) with an average of 20 clones/pseudo-molecule (Table 5). These mapped MTP clones coming from different contigs of the physical map represent ~70 Mb genome of physical map integrated into draft genome and genetic map. The low number of BACs mapped on the reference genome during the present study may be due to the following: (i) exclusion of 37,230 BES having different types of repeat elements; (ii) exclusion of BAC clones, where paired BESs aligned <100 and >200 kb apart; (iii) exclusion of majority of BACs showing multiple region mapping on either the same pseudo-molecule or different pseudo-molecules; and (iv) exclusion of several paired BES from mapping showing orientation in either the same or opposite directions indicating potential inversions. Similar type of potential inversions, repeat elements, mapping on different regions on pseudo-molecules have been reported earlier during the in silico mapping and integration of physical maps with the genome sequence in rice (Katagiri et al. 2004), tomato (Asamizu et al. 2012) and soybean (Ha et al. 2012). The removal of 37,230 repeat BES from chromosomal mapping was aimed to avoid the complications of map construction. Integration of the physical map of a Desi variety during the present study with the draft genome sequence of Kabuli chickpea will help in locating chromosomal structural arrangements between Desi (ICC 4958) and Kabuli (CDS Frontier) genotypes in future studies.
Candidate stress-responsive genes
Among 654 genes present in the “QTL-hotspot”, the GO annotation-indicated genes like ERECTA-like kinase have a role in drought tolerance, and it has been reported that this gene enhances transpiration efficiency (Masle et al. 2005) and water use efficiency and transpiration efficiency in Arabidopsis (Xing et al. 2011). Several other genes like thiamine thiazole synthase and cysteine-rich receptor-like protein kinase, calmodulin-binding heat shock protein as well as other heat shock transcription factors, tubulin beta-chain, have roles in adaptation to various stress conditions. In addition, genes like fructose-bisphosphate aldolase, transmembrane protein which respond to salt stress as well as CRT/DRE-binding factor 4 gene which has shown a prominent role in abiotic stress response in Medicago (Li et al. 2011) were also found in this region, indicating the potential of this region to enhance the tolerance to several abiotic stresses.
In the case of Ascochyta blight resistance QTL region, among 306 genes, genes like BED finger-nbs resistance protein and gene with leucine-rich repeat domain are typically involved in host resistance mechanism like DNA-directed RNA polymerase subunit beta, receptor-like protein kinase and Ser-Thr protein kinase. Further, this region also harbours NAC domain protein for systemic acquired resistance as well as NB-LRR-type disease resistance protein Rps1-k-2 and ABC-type transport system involved in resistance to organic solvents’ periplasmic component.
Similarly, in the case of Fusarium wilt resistance QTL region, genes that have shown their activity in defense response to bacterium in Medicago like pheophorbide A oxygenase were found. This region also harbours superoxide dismutase which has shown differential expression in WR 315 compared to JG 62 in infected state as well as zinc finger protein and translation initiation factor (Gupta et al. 2013).
These candidate genes may be validated by using some functional genomics approaches like Targeting Induced Local Lesions IN Genome (TILLING), quantitative RT-PCR, over expression, etc., using the appropriate genetic material. In summary, integration of the physical map with the genetic maps and genome sequence of chickpea should help the international chickpea community for cloning of genes related to important tolerance/resistance to abiotic and biotic stresses as well as agronomic traits.
References
Abu-Salem FM, Abou EA (2011) Physico-chemical properties of tempeh produced from chickpea seeds. Arab J Am Sci 7:107–118
Anbessa Y, Taran B, Warkentin TD, Tullu A, Vandenberg A (2009) Genetic analyses and conservation of QTL for Ascochyta blight resistance in chickpea. Theor Appl Genet 119:757–765
Anuradha C, Gaur PM, Pande S, Gali KK, Ganesh M, Kumar J, Varshney RK (2011) Mapping QTL for resistance to Botrytis grey mould in chickpea. Euphytica 182:1–9
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Report 9:208–218
Aryamanesh N, Nelson MN, Yan G, Clarke HJ, Siddique KHM (2010) Mapping a major gene for growth habit and QTLs for Ascochyta blight resistance and flowering time in a population between chickpea and Cicer reticulatum. Euphytica 173:307–319
Asamizu E, Shirasawa K, Hirakawa H, Sato S, Tabata S, Yano K, Ariizumi T, Shibata D, Ezura H (2012) Mapping of micro-Tom BAC-end sequences to the reference tomato genome reveals possible genome rearrangements and polymorphisms. Int J Plant Genomics. doi:10.1155/2012/437026
Bakker E, Butterbach P, Rouppe van der Voort JN, van der Vossen EA, van Vliet J, Bakker J, Goverse A (2003) Genetic and physical mapping of homologues of the virus resistance gene Rx1 and the cyst nematode resistance gene Gpa2 in potato. Theor Appl Genet 106:1524–1531
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Chamarthi SK, Kumar A, Vuong T, Blair MW, Gaur PM, Nguyen HT, Varshney RK (2011) Trait mapping and molecular breeding in legumes: concepts and examples in soybean, common bean and chickpea. In: Pratap A, Kumar J (eds) Biology and breeding of food legumes. CABI International, Oxfordshire, pp 296–313
Chen M et al (2002) An integrated physical and genetic map of the rice genome. Plant Cell 14:537–545
Choudhary S, Gaur R, Gupta S, Bhatia S (2012) EST-derived genic-molecular markers: development and utilization for generating an advanced transcript map of chickpea. Theor Appl Genet 124:1449–1462
Cobos M, Rubio J, Fernandez-Romero M, Garza R, Moreno M, Millán T, Gil J (2007) Genetic analysis of seed size, yield and days to flowering in a chickpea RIL population derived from a Kabuli × Desi cross. Ann Appl Biol 151:33–42
Cuc LM, Mace E, Crouch J, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:55
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Garg R, Patel RK, Jhanwar S, Priya P, Bhattacharjee A, Yadav G, Bhatia S, Chattopadhyay D, Tyagi AK, Jain M (2011) Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol 156:1661–1678
Gaur R, Azam S, Jeena G, Khan AW, Choudhary S, Jain M, Yadav G, Tyagi AK, Chattopadhyay D, Bhatia S (2012) High-Throughput SNP discovery and genotyping for constructing a saturated linkage map of chickpea (Cicer arietinum L.). DNA Res. doi:10.1093/dnares/dss018
Gregory SG, Howell GR, Bentley DR (1997) Genome mapping by fluorescent fingerprinting. Genome Res 7:1162–1168
Gu YQ, Ma Y, Huo N, Vogel JP, You FM, Lazo GR, Nelson WM, Soderlund C, Dvorak J, Anderson OD, Luo M-C (2009) A BAC-based physical map of Brachypodium distachyon and its comparative analysis with rice and wheat. BMC Genomics 10:496
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, Bhanu Prakash A, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122:1577–1589
Gupta S, Bhar A, Chatterjee M, Das S (2013) Fusarium oxysporum f. sp. ciceri race 1 induced redox state alterations are coupled to downstream defense signaling in root tissues of chickpea (Cicer arietinum L.). PLoS One 8:e73163
Ha J, Abernathy B, Nelson W, Grant D, Wu X, Nguyen HT, Stacey G, Yu Y, Wing RA, Shoemaker RC, Jackson SA (2012) Integration of the draft sequence and physical map as a framework for genomic research in soybean (Glycine max (L.) Merr.) and wild soybean (Glycine soja Sieb. and Zucc.). Genes Genomes Genet 2:321–329
Han Y, Zheng D, Vimolmangkang S, Khan MA, Beever JE, Korban SS (2011) Integration of physical and genetic maps in apple confirms whole-genome and segmental duplications in the apple genome. J Exp Bot 62:5117–5130
Hillier LW et al (2008) Whole-genome sequencing and variant discovery in C. elegans. Nat Methods 5:183–188
Hiremath PJ et al (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732
Huo N et al (2008) The nuclear genome of Brachypodium distachyon: analysis of BAC end sequences. Funct Integr Genomics 8:135–147
Islam-Faridi MN, Childs KL, Klein PE, Hodnett G, Menz MA, Klein RR, Rooney WL, Mullet JE, Stelly DM, Price HJ (2002) A molecular cytogenetic map of sorghum chromosome 1. Fluorescence in situ hybridization analysis with mapped bacterial artificial chromosomes. Genetics 161:345–353
Jain M et al (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. doi:10.1111/tpj.12173
Katagiri S, Wu J, Ito Y, Karasawa W, Shibata M, Kanamori H, Katayose Y, Namiki N, Matsumoto T, Sasaki T (2004) End sequencing and chromosomal in silico mapping of BAC clones derived from an indica rice cultivar, Kasalath. Breed Sci 54:273–279
Kelleher CT et al (2007) A physical map of the highly heterozygous Populus genome: integration with the genome sequence and genetic map and analysis of haplotype variation. Plant J 50:1063–1078
Kidd JM et al (2008) Mapping and sequencing of structural variation from eight human genomes. Nature 453:56–64
Klein PE et al (2000) A high-throughput AFLP based method for constructing integrated genetic and physical maps: progress towards a sorghum genome map. Genome Res 10:789–807
Kottapalli P, Gaur PM, Katiyar SK, Crouch JH, Buhariwalla HK, Pande S, Gali KK (2009) Mapping and validation of QTLs for resistance to an Indian isolate of Ascochyta blight pathogen in chickpea. Euphytica 165:79–88
Kudapa H et al (2014) Comprehensive transcriptome assembly of chickpea (Cicer arietinum L.) using Sanger and next generation sequencing platforms: development and applications. PLoS One 9:e86039. doi:10.1371/journal.pone.0086039
Lewin HA, Larkin DM, Pontius J, O’Brien SJ (2009) Every genome sequence needs a good map. Genome Res 19:1925–1928
Li D, Zhang Y, Hu X, Shen X, Ma L, Su Z, Wang T, Dong J (2011) Transcriptional profiling of Medicago truncatula under salt stress identified a novel CBF transcription factor MtCBF4 that plays an important role in abiotic stress responses. BMC Plant Biol 11:109
Lichtenzveig J, Scheuring C, Dodge J, Abbo S, Zhang HB (2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theor Appl Genet 110:492–510
Luo M-C, Thomas C, You FM, Hsiao J, Ouyang S, Buell CR, Malandro M, McGuire PE, Anderson OD, Dvorak J (2003) High-throughput fingerprinting of bacterial artificial chromosomes using the snapshot labeling kit and sizing of restriction fragments by capillary electrophoresis. Genomics 82:378–389
Marra MA, Kucaba TA, Dietrich NL, Green ED, Brownstein B, Wilson RK, McDonald KM, Hillier LW, McPherson JD, Waterston RH (1997) High throughput fingerprint analysis of large-insert clones. Genome Res 7:1072–1084
Masle J, Gilmore SR, Farquhar GD (2005) The ERECTA gene regulates plant transpiration efficiency in Arabidopsis. Nature 436:866–870
Meinke D, Sweeney C, Muralla R (2009) Integrating the genetic and physical maps of Arabidopsis thaliana: identification of mapped alleles of cloned essential (emb) genes. PLoS One 4(10):e7386
Millán T et al (2010) A consensus genetic map of chickpea (Cicer arietinum L.) based on 10 mapping populations. Euphytica 175:175–189
Nayak SN et al (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120:1415–1441
Nelson WM, Bharti AK, Butler E, Wei F, Fuks G, Kim H, Wing RA, Messing J, Soderlund C (2005). Whole-genome validation of high-information-content fingerprinting. Plant Physiol 139: 27–33
Rounsley S et al (2009) De novo next generation sequencing of plant genomes. Rice 2:35–43
Sabbavarapu MM et al (2013) Molecular mapping of QTLs for resistance to Fusarium wilt (race 1) and Ascochyta blight in chickpea (Cicer arietinum L.). Euphytica 193:121–133
Sasaki T et al (2005) The map-based sequence of the rice genome. Nature 436:793–800
Sato S et al. (2008) Genome structure of the legume, Lotus japonicus. DNA Res 15:227–239
Schmutz J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M (1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci 89:8794–8797
Tao Q, Wang A, Zhang H-B (2002) One large-insert plant transformation-competent BIBAC library and three BAC libraries of Japonica rice for genome research in rice and other grasses. Theor Appl Genet 105:1058–1066
Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinforma 25:4.10.1–4.10.14
Tekeoglu M, Rajesh PN, Muehlbauer FJ (2002) Integration of sequence tagged a microsatellite sites to the chickpea genetic map. Theor Appl Genet 105:47–854
Thudi M et al (2011) Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS One 6(11):e27275
Thudi M, Li Y, Jackson SA, May GD, Varshney RK (2012) Current state-of-art of sequencing technologies for plant genomics research. Brief Funct Genomics 11:3–11
Udupa SM, Baum M (2003) Genetic dissection of pathotype-specific resistance to Ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106:1196–1202
Upadhyaya HD, Thudi M, Dronavalli N, Gujaria N, Singh S, Sharma S, Varshney RK (2011) Genomic tools and germplasm diversity for chickpea improvement. Plant Genet Res 9:45–48
Vadez V, Krishnamurthy L, Thudi M, Colmer TD, Turner NC, Siddique KHM, Gaur PM, Varshney RK (2012) Assessment of ICCV 2 × JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTLs for seed yield and seed number. Mol Breed 30:9–21
van der Vossen EA, van der Voort JN, Kanyuka K, Bendahmane A, Sandbrink H, Baulcombe DC, Bakker J, Stiekema WJ, Klein-Lankhorst RM (2000) Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: a virus and a nematode. Plant J 23:567–576
Varshney RK et al (2009) A comprehensive resource of drought- and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genomics 10:523
Varshney RK, Thudi M, May GD, Jackson SA (2010) Legume genomics and breeding. Plant Breed Rev 33:257–304
Varshney RK et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30:83–89
Varshney RK et al (2013a) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Varshney RK, Ribaut J-M, Buckler ES, Tuberosa R, Rafalski JA, Langridge P (2013b) Can genomics boost productivity of orphan crops? Nat Biotechnol 30:1172–1176
Varshney RK et al (2014) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127:445–462
Wei F et al (2007) Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 3:e123. doi:10.1371/journal.pgen.0030123
Wheeler DA et al (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452:872–876
Wu C, Sun S, Nimmakayala P, Santos FA, Meksem K, Springman R, Ding K, Lightfoot DA, Zhang HB (2004) A BAC- and BIBAC-based physical map of the soybean genome. Genome Res 14:319–326
Xing HT, Guo P, Xia XL, Yin WL (2011) PdERECTA, a leucine-rich repeat receptor-like kinase of poplar, confers enhanced water use efficiency in Arabidopsis. Planta 234:229–241
Young ND et al (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480:520–524
Yu Q et al (2009) A physical map of the papaya genome with integrated genetic map and genome sequence. BMC Genomics 10:371
Zhang X et al (2010) A BAC/BIBAC-based physical map of chickpea, Cicer arietinum L. BMC Genomics 11:501
Zhebentyayeva TN et al (2008) A framework physical map for peach, a model Rosaceae species. Tree Genet Genomes 4:745–756
Acknowledgement
The authors are thankful to Douglas R. Cook, University of California, Davis, USA, and Aamir W. Khan for their help during analysis and interpretation of data. This study was supported as a part of the Tropical Legumes I (TLI- Objective 4: Chickpea) project funded by Bill & Melinda Gates Foundation (BMGF) through CGIAR Generation Challenge Programme (RKV) and National Institute of Plant Genome Research (NIPGR), Department of Biotechnology, Government of India (SB). This work has been undertaken as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of CGIAR Consortium.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Comparison of sequence, physical and genetic maps. a-h are comparisons of each linkage group with respective psuedomolecules (JPEG 751 kb)
(JPEG 646 kb)
(JPEG 613 kb)
(JPEG 845 kb)
(JPEG 854 kb)
(JPEG 892 kb)
(JPEG 652 kb)
(JPEG 440 kb)
Supplementary Table 1
Markers mapped on to physical map and associated trait QTLs (DOCX 85 kb)
Supplementary Table 2
The distribution of repetitive sequences in 5,3316 BES used for their mapping on chickpea genome sequence (DOCX 17 kb)
Supplementary Table 3
List of genes in drought tolerance "QTL-hotspot" region (XLSX 46 kb)
Supplementary Table 4
List of genes in Ascochyta blight resistance QTL region (XLSX 30 kb)
Supplementary Table 5
List of genes in Fusarium wilt resistance QTL region (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Varshney, R.K., Mir, R.R., Bhatia, S. et al. Integrated physical, genetic and genome map of chickpea (Cicer arietinum L.). Funct Integr Genomics 14, 59–73 (2014). https://doi.org/10.1007/s10142-014-0363-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-014-0363-6