
Abstract
We present clinical practice recommendations for the treatment of children with Alport syndrome who are not enrolled in clinical trials. Our goal is to promote early initiation of a standard therapeutic approach that will facilitate assessment of the safety and efficacy of the protocol. The treatment protocol is based on the reduction of proteinuria, intraglomerular pressure, and renal fibrosis via interference with the renin–angiotensin–aldosterone system.
Similar content being viewed by others

Introduction
Investigation of Alport syndrome (AS) has elucidated the roles of type IV collagen in basement membranes and described the consequences of type IV collagen mutations for renal, cochlear, and ocular structure and function. Research studies employing animal models of AS have suggested interventions that may delay or reverse the renal effects of type IV collagen mutations, but these approaches have yet to be prospectively examined in human subjects with the disease. While we wait for clinical trials to be organized, funded, and carried to completion, children and adults with AS continue to progress towards end-stage renal disease (ESRD). What can nephrologists offer in the meantime, and what can we learn from clinical practice about the efficacy and safety of common treatment approaches?
The Alport Syndrome Research Collaborative, currently comprising investigators in Canada, China, France, Germany, and the United States, has developed clinical practice recommendations aimed at standardizing therapy for people with AS who are not enrolled in clinical trials. It is our hope that adoption of a consistent approach to therapy will allow pooling and analysis of data about treatment responses and efficacy.
Disclaimer
These are general clinical practice recommendations for off-label therapy with inhibitors of the renin–angiotensin–aldosterone system (RAAS) and may not be appropriate for all children with AS. It is expected that practitioners will carefully consider a child’s baseline renal function in applying these guidelines, will appropriately monitor for adverse effects of therapy, and will adjust or discontinue therapy as needed.
Natural history of untreated Alport syndrome
Genetics and genotype-phenotype correlations
Alport syndrome can be transmitted as an X-linked, autosomal recessive or autosomal dominant disorder. About 80% of individuals with AS have X-linked disease (XLAS) due to mutations in the COL4A5 gene. Males with XLAS progress inexorably to ESRD, with ESRD risks of 50% by age 25, 90% by age 40, and nearly 100% by age 60 [1]. Age at ESRD is strongly correlated with COL4A5 genotype in males with XLAS [1–3]; risk of ESRD by age 30 is 90% for deletions and nonsense mutations of COL4A5, 70% for splicing mutations, and 50% for missense mutations. The known strong genotype-phenotype correlation provides a rationale for using COL4A5 genotype data to guide the timing and intensity of intervention. In most families with XLAS, age at ESRD is fairly similar among affected males. In the absence of COL4A5 genotype data, timing of ESRD can be predicted for a young affected male on the basis of ESRD timing in older affected male relatives.
The effects of COL4A5 genotype on age at ESRD are not observed in females with XLAS, likely due to the overwhelming influence of X-inactivation [4]. In XLAS females, the timing and intensity of intervention should be guided by risk factors for progression to ESRD: proteinuria, gross hematuria, and hearing loss [5, 6].
Autosomal recessive AS (ARAS) accounts for about 15% of individuals with the disease and arises from mutations in both alleles of either COL4A3 or COL4A4. Genotype-phenotype data for ARAS is relatively sparse. In general, individuals with ARAS carry a high risk of ESRD by age 30. Only about 5% of individuals with AS have autosomal dominant disease (ADAS). As ADAS tends to progress at a relatively slow velocity [7], there is less urgency to consider initiation of intervention in childhood.
Clinicopathological correlations
In general, AS is characterized by genetically determined dysfunction of the glomerular filter, mainly caused by mutations in the collagens assembling the glomerular basement membrane (GBM). In consequence, the earliest sign of GBM filter dysfunction is hematuria followed by albuminuria and subsequent nonselective proteinuria in increasing magnitude. Ultimately, not the GBM damage per se but the pro-inflammatory and pro-fibrotic consequences both in the tubulointerstitium and in the glomeruli resulting from progressive proteinuria, eventually lead to the development of ESRD [8].
Overt proteinuria is typically absent in infant males with XLAS. Age at identification of overt proteinuria shows interfamilial variability and ranges from early childhood to adolescence. In dogs with XLAS, a period of microalbuminuria precedes the development of overt proteinuria and quantitative increases in interstitial volume due to tubular atrophy and fibrosis [9]. Dogs with XLAS exhibit increased proximal tubular epithelial cell uptake of albumin, a process that has been linked to cellular injury [9]. Preliminary data from the Alport Syndrome Treatments and Outcomes Registry (ASTOR) indicates that boys with AS also exhibit a transitional stage of microalbuminuria before overt proteinuria becomes established (manuscript in preparation). It has yet to be demonstrated that suppression of microalbuminuria has anti-fibrotic effects in AS, although this is a reasonable hypothesis.
Measurements of cortical interstitial volumes in AS males have shown that interstitial fibrosis is unusual before age 10 [10]. Cortical interstitial volumes become abnormal in many AS males during the second decade of life, and are inversely correlated with glomerular filtration rates [10]. These observations suggest that, (1) as in other chronic glomerulopathies, interstitial fibrosis is a significant contributor to loss of renal function, and (2) prevention of interstitial fibrosis in AS males may require intervention during childhood.
Therapeutic studies
Several therapies improve outcomes in animal models of AS, including angiotensin-converting enzyme (ACE) inhibition [11, 12], AT1-receptor blockade (ARB) [11, 12], inhibitors of TGF-β1, matrix metalloproteinases, vasopeptidase A or HMG-CoA reductase [13–16]; chemokine receptor 1 blockade [17], BMP-7 [18], stem cells [19–22], and irradiation [23]. However, none of these approaches has been prospectively studied in human AS populations.
In ARAS mice, initiation of ACE inhibitor therapy before onset of proteinuria suppressed proteinuria and azotemia and doubled length of survival [11], a therapeutic benefit that has yet to be exceeded by any other intervention. A smaller but still significant improvement in outcome was achieved when ACE inhibition was started after onset of proteinuria [11]. ACE inhibitor therapy begun before onset of proteinuria lengthened survival in canine XLAS [24].
Retrospective analysis of registry data strongly suggests that ACE inhibition delays ESRD and improves life expectancy in AS patients in a time-dependent manner [25]. This observation highlights the importance of early and accurate diagnosis of AS, including specific genotyping whenever feasible (for information on diagnostic methods in AS, please see www.genereviews.org). Besides the beneficial effects of RAAS blockade on outcomes in experimental and human AS, there are additional compelling reasons to recommend RAAS blockade in AS patients until a superior therapy is identified. First, ACE inhibitor therapy at doses that achieve suppression of proteinuria has been used with a high degree of safety in children with chronic kidney disease [26, 27]. Further, these agents are widely available and relatively inexpensive, making this therapy accessible to AS patients worldwide.
Clinical trials
We recognize that a randomized clinical trial would be the best way to evaluate the efficacy of ACE inhibition or any other intervention in children with AS. We also recognize that widespread adoption of our recommendations may reduce the pool of children who are eligible for clinical trials of potential therapies for AS. Nevertheless, we encourage eligible subjects and their families to consider participation in clinical trials wherever and whenever possible. For example, by the time this document is published, a clinical trial of ACE inhibition in AS will be recruiting subjects residing in Germany, or willing and able to travel to Germany (contact: studie@alport.de; for information and to download flyer see www.alport.de/EARLY_PRO-TECT). The goal of this trial is to clarify if an early start of therapy (in patients with isolated hematuria or microalbuminuria) delays renal failure even more effectively than later onset of therapy (in proteinuric patients) and above all if therapy at early stages of AS is safe.
For those subjects unable to take part in clinical trials, we believe that valuable efficacy and safety data can be collected if these subjects are treated according to a standardized protocol. Currently, many children with AS are receiving ACE inhibitor therapy in a non-standardized fashion, and information about treatment responses is not being centrally collected or analyzed. We believe that this represents a wasted opportunity.
Treatment recommendations
Retrospective registry data strongly suggests that in subjects with AS ACE inhibitor therapy initiated once proteinuria has developed but while glomerular filtration rate is well preserved delays ESRD [25]. Similarly, in mice and dogs with AS, interventions that delay the onset of proteinuria or reduce established proteinuria prolong renal survival [11, 24]. Based on these observations, and on the general consensus that suppression of intraglomerular pressure and proteinuria is an important component of the management of chronic glomerular diseases, treatment of proteinuric AS patients with medications that diminish proteinuria is recommended. Proteinuria in children is defined as urine protein-creatinine ratio persistently greater than 0.2 mg/mg in children over 2 years of age, or urinary protein excretion greater than 4 mg/m2/h in a timed collection [28, 29].
Cortical interstitial volume fraction (VvI/C) is a measure of interstitial fibrosis and tubular atrophy [30]. There is a strong inverse correlation between VvI/C and creatinine clearance in boys with AS [10]. In these boys, VvI/C is typically within the normal range during the first decade of life, when creatinine clearance is also normal, but often begins to increase during adolescence concurrent with declining creatinine clearance. We presume that COL4A5 mutations associated with relatively rapid progression to ESRD, such as deletions, nonsense, and splicing mutations, result in earlier onset and more aggressive development of interstitial fibrosis and tubular atrophy. In dogs and mice with AS, the onset of proteinuria precedes measurable increases in interstitial fibrosis. In light of these observations, we recommend that in boys with AS who have deletion, nonsense or splicing mutations, or who have a family history of ESRD before age 30, monitoring of urine protein excretion should begin early in life and that an aggressive approach to initiating and escalating proteinuria-suppressing therapies should be followed.
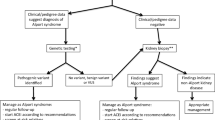
Based on these principles, we make the following recommendations aimed at preventing renal tubular epithelial cell injury and suppressing fibrogenic processes in the renal interstitium (see Fig. 1):

Algorithm for identifying children with familial hematuria (Alport syndrome or hematuria with thin glomerular basement membranes) who are candidates for intervention. IHC immunohistochemistry; EM electron microscopy; GBM glomerular basement membrane *Depending upon availability and local practice
-
1.
Monitoring for microalbuminuria and proteinuria should be initiated by age 1 year in at risk children, or as soon as a diagnosis of Alport syndrome is established, and repeated at least annually.
-
2.
Affected individuals with overt proteinuria (urine protein-creatinine ratio persistently greater than 0.2 mg/mg, or urinary protein excretion greater than 4 mg/m2/h in a timed collection) should receive treatment.
-
3.
Treatment should be considered in affected boys with microalbuminuria in whom the risk of ESRD by age 30 is high, such as those with COL4A5 deletions, nonsense or splicing mutations, or a history of ESRD before age 30 in affected male relatives (Table 1). We recognize that access to molecular genetic testing for Alport syndrome, and coverage by insurers, is variable. We recommend that the Alport genotype be determined whenever feasible, to facilitate identification of those at high risk of ESRD by age 30.
Table 1 Recommendations for intervention based on urinary findings and anticipated disease course
Target
The optimal target for lowering of urine protein levels is uncertain. Our recommendations are based upon an arbitrary goal of a urine protein:creatinine ratio of less than 0.5 mg/mg if the baseline value is greater than 1.0 mg/mg, or a 50% reduction if the baseline value is greater than 0.2 but less than 1.0. When therapy is initiated in subjects with microalbuminuria, we recommend a target microalbumin:creatinine ratio of less than 50–100 mg/g creatinine.
Proteinuria may persist at levels that exceed these targets, despite maximum dosing of first- and second-line agents. In these cases, we recommend continuing therapy, with adjustment of dosing as indicated by growth and by renal function.
Agents
First line
We chose angiotensin-converting enzyme (ACE) inhibition as first-line therapy for several reasons. First, ACE inhibition is the choice of most nephrologists for initial non-immunologic therapy of proteinuric glomerular disease. Consequently, practitioners have extensive experience with dosing these agents and are familiar with their adverse effects. ACE inhibitors are widely available and relatively inexpensive. The Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial demonstrated that ACE inhibition with ramipril is associated with very low frequencies of adverse events in children with chronic kidney disease and at least transient reductions in proteinuria [26, 27]. Because of the ESCAPE experience, we chose ramipril as the reference ACE inhibitor, and suggest equivalent doses of other ACE inhibitors in Table 2. Finally, ramipril therapy started before or after onset of proteinuria significantly prolonged survival in mice with autosomal recessive Alport syndrome, and its effects were superior to those of candesartan [11, 12].
In the ESCAPE trial, reduction in proteinuria and preservation of glomerular filtration rate were correlated with ramipril’s antihypertensive effect [26, 27]. Many of the Alport subjects in whom therapy is initiated when they have developed microalbuminuria or mild proteinuria will be normotensive. If the renoprotective properties of ACE inhibitors are primarily due to their antihypertensive effects, the impact on Alport subjects may be insignificant. However, ramipril delays proteinuria and prolongs survival in normotensive autosomal recessive Alport syndrome mice, indicating that other effects of ACE inhibition are important in this model [11, 12].
Second line
We propose two alternatives for second-line therapy, angiotensin receptor blockade (ARB) and aldosterone inhibition. For those nephrologists with experience combining an ACE inhibitor with an ARB, ARB may be the more comfortable, familiar approach. We suggest losartan as the reference ARB, based on published experience in children with chronic kidney disease [31]. In a recent study of 30 proteinuric children with AS, losartan was found to reduce proteinuria to a greater extent than placebo or amlodipine [32]. We recommend a relatively low starting dose, given that the ARB will be added to ACE inhibition (Table 3). If an ARB is used instead of an ACE inhibitor, for example because a patient tolerates ACE inhibition poorly, a higher starting dose for the ARB would be appropriate.
There are a limited number of published reports regarding the use of combination therapy with an ACE inhibitor and an ARB in children with chronic proteinuric renal diseases. Ten children with chronic kidney disease and persistent proteinuria, despite maximal doses of an ACE inhibitor, exhibited a sustained reduction in proteinuria after addition of losartan, with no significant changes in blood pressure or glomerular filtration rate [33]. In another small study of ten children with chronic kidney disease and proteinuria that employed a cross-over design, combination therapy with an ACE inhibitor and an ARB reduced proteinuria to a significantly greater extent than either agent alone [34].
In a small group of patients with AS, combined therapy with an ACE inhibitor and spironolactone suppressed proteinuria to a greater extent than the combination of an ACE inhibitor with an ARB [35]. Hyperkalemia was not encountered in this small group of patients. Increased aldosterone levels (aldosterone escape) may contribute to persistent proteinuria in Alport patients receiving ACE inhibitor therapy [36]. Aldosterone inhibition could be used as the initial second-line agent, or as an alternative to ineffective ARB therapy (Table 4).
Potential adverse effects of therapy
Potential adverse effects of the therapeutic approach described above include orthostatic hypotension, fetopathy in ovulating females, hyperkalemia, reversible decline in glomerular filtration rate, and gynecomastia. In the ESCAPE trial, only three of 352 children who received ramipril for at least 6 months required discontinuation of therapy because of orthostatic hypotension or hyperkalemia [24]. ACE inhibitors and ARBs should be used with caution in ovulating females in order to avoid fetal injury.
In the ONgoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) study, the combination of ramipril and telmisartan resulted in increased risk of adverse renal outcomes (doubling of serum creatinine and requirement for dialysis), hypotensive symptoms, syncope, and hyperkalemia, compared to ramipril monotherapy [37]. Subjects who received combination therapy also had an increased risk of mortality, although not statistically significant [37]. The relevance of the results of the ONTARGET study, in which study subjects had a mean age of 66 years and were at high risk for cardiovascular events due to vascular disease or diabetes, to children and young adults with Alport syndrome is unclear.
Chronic administration of spironolactone is associated with an increased incidence of serum potassium greater than 5.5 mEq/l (about 3% of subjects participating in clinical trials), gynecomastia (about 5% of patients), and acute renal insufficiency (1–5% of subjects) [38]. We recommend regular monitoring of serum potassium and creatinine levels in treated Alport patients, especially those receiving combination therapy.
Reporting of treatment experience
We are in the process of developing an online reporting tool that will allow practitioners to upload de-identified information regarding treatment responses to a secure site. Once the tool is in place, we will assist interested practitioners in obtaining local IRB approval for participation. We will use listservs and blast e-mails from the Alport Syndrome Treatments and Outcomes Registry to announce the availability of the site. In the meantime, we encourage practitioners to share treatment experiences via e-mail with Dr. Kashtan.
References
Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer K-O, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schroder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC (2000) X-linked Alport syndrome: natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol 11:649–657
Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M (2002) Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counseling. Nephrol Dial Transpl 17:1218–1227
Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, Schrier RW (2010) Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 21:876–883
Rheault MN, Kren SM, Hartich LA, Wall M, Thomas W, Mesa HA, Avner P, Lees GE, Kashtan CE, Segal Y (2010) X-inactivation modifies disease severity in female carriers of murine X-linked Alport syndrome. Nephrol Dial Transplant 25:764–769
Grunfeld J-P, Noel LH, Hafez S, Droz D (1985) Renal prognosis in women with hereditary nephritis. Clin Nephrol 23:267–271
Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schroder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC (2003) X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol 14:2603–2610
Pochet JM, Bobrie G, Landais P, Goldfarb B, Grunfeld J-P (1989) Renal prognosis in Alport’s and related syndromes: influence of the mode of inheritance. Nephrol Dial Transpl 4:1016–1021
Gross O, Kashtan CE (2009) Treatment of Alport syndrome: beyond animal models. Kidney Int 76:599–603
Vinge L, Lees GE, Nielsen R, Kashtan CE, Bahr A, Christensen EI (2010) The effect of progressive glomerular disease on megalin-mediated endocytosis in the kidney. Nephrol Dial Transplant 25:2458–2467
Kashtan CE, Gubler MC, Sisson-Ross S, Mauer M (1998) Chronology of renal scarring in males with Alport syndrome. Pediatr Nephrol 12:269–274
Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M (2003) Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int 63:438–446
Gross O, Schulze-Lohoff E, Koepke ML, Beirowski B, Addicks K, Bloch W, Smyth N, Weber M (2004) Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol Dial Transplant 19:1716–1723
Sayers R, Kalluri R, Rodgers KD, Shield CF, Meehan DT, Cosgrove D (1999) Role for transforming growth factor-beta 1 in Alport renal disease progression. Kidney Int 56:1662–1673
Gross O, Koepke ML, Beirowski B, Schulze-Lohoff E, Segerer S, Weber M (2005) Nephroprotection by antifibrotic and anti-inflammatory effects of the vasopeptidase inhibitor AVE7688. Kidney Int 68:456–463
Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF, Werb Z, Kalluri R (2006) Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med 3:e100
Koepke ML, Weber M, Schulze-Lohoff E, Beirowski B, Segerer S, Gross O (2007) Nephroprotective effect of the HMG-CoA-reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome. Nephrol Dial Transplant 22:1062–1069
Ninichuk V, Gross O, Reichel C, Kandoga A, Pawar RD, Ciubar R, Segerer S, Belemezova E, Radomska E, Luckow B, Perez de Lema G, Murphy PM, Gao J, Henger A, Kretzler M, Horuk R, Weber M, Krombach F, Schlondorff D, Anders H (2005) Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 16:977–985
Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, Kalluri R (2003) Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol 285:F1060–F1067
Ninichuk V, Gross O, Segerer S, Hoffmann R, Radomska E, Buchstaller A, Huss R, Akis N, Schlondorff D, Anders HJ (2006) Multipotent mesenchymal stem cells reduce interstitial fibrosis but do not delay progression of chronic kidney disease in collagen4A3-deficient mice. Kidney Int 70:121–129
Sugimoto H, Mundel TM, Sund M, Xie L, Cosgrove D, Kalluri R (2006) Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci USA 103:7321–7326
Gross O, Borza DB, Anders HJ, Licht C, Weber M, Segerer S, Torra R, Gubler MC, Heidet L, Harvey S, Cosgrove D, Lees G, Kashtan C, Gregory M, Savige J, Ding J, Thorner P, Abrahamson DR, Antignac C, Tryggvason K, Hudson B, Miner JH (2009) Stem cell therapy for Alport syndrome: the hope beyond the hype. Nephrol Dial Transplant 24:731–734
Lebleu V, Sugimoto H, Mundel TM, Gerami-Naini B, Finan E, Miller CA, Gattone VH, Lu L, Shield CF, Folkman J, Kalluri R (2009) Stem cell therapies benefit Alport syndrome. J Am Soc Nephrol 20:2359–2370
Katayama K, Kawano M, Naito I, Ishikawa H, Sado Y, Asakawa N, Murata T, Oosugi K, Kiyohara M, Ishikawa E, Ito M, Nomura S (2008) Irradiation prolongs survival of Alport mice. J Am Soc Nephrol 19:1692–1700
Grodecki KM, Gains MJ, Baumal R, Osmond DH, Cotter B, Valli VE, Jacobs RM (1997) Treatment of X-linked hereditary nephritis in Samoyed dogs with angiotensin-converting enzyme inhibitor. J Comp Pathol 117:209–225
Gross O, Licht C, Anders H, Hoppe B, Beck B, Tonshoff B, Hocker B, Wygoda S, Ehrich J, Pape L, Konrad M, Rascher W, Dotsch J, Muller-Wiefel D, Hoyer P, Knebelmann B, Pirson Y, Grunfeld J, Niaudet P, Cochat P, Heidet L, Lebbah S, Torra R, Friede T, Lange K, Muller GA, Weber M (2012) Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. doi:10.1038/ki.2011.407
Wuhl E, Mehls O, Schaefer F (2004) Antihypertensive and antiproteinuric efficacy of ramipril in children with chronic renal failure. Kidney Int 66:768–776
Wuhl E, Trivelli A, Picca S, Litwin M, Peco-Antic A, Zurowska A, Testa S, Jankauskiene A, Emre S, Caldas-Afonso A, Anarat A, Niaudet P, Mir S, Bakkaloglu A, Enke B, Montini G, Wingen AM, Sallay P, Jeck N, Berg U, Caliskan S, Wygoda S, Hohbach-Hohenfellner K, Dusek J, Urasinski T, Arbeiter K, Neuhaus T, Gellermann J, Drozdz D, Fischbach M, Moller K, Wigger M, Peruzzi L, Mehls O, Schaefer F (2009) Strict blood-pressure control and progression of renal failure in children. N Engl J Med 361:1639–1650
Elises JS, Griffiths PD, Hocking MD, Taylor CM, White RH (1998) Simplified quantification of urinary protein excretion in children. Clin Nephrol 30:225–229
Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J (2000) Evaluation and management of proteinuria and nephrotic syndrome in children: recommendations from a pediatric nephrology panel established at the National Kidney Foundation Conference on Proteinuria, Albuminuria, Risk, Assessment, and Elimination (PARADE). Pediatrics 105:1242–1249
Lane PH, Steffes MW, Fioretto P, Mauer SM (1993) Renal interstitial expansion in insulin-dependent diabetes mellitus. Kidney Int 43:661–667
Ellis D, Moritz M, Vats A, Janosky JE (2004) Antihypertensive and renoprotective efficacy and safety of losartan. Am J Hypertens 17:928–935
Webb NJ, Lam C, Shahinfar S, Strehlau J, Wells TG, Gleim GW, Le Bailly De Tilleghem C (2011) Efficacy and safety of losartan in children with Alport syndrome – results from a subgroup analysis of a prospective, randomized placebo- or amlodipine-controlled trial. Nephrol Dial Transplant 28:2521–2526
Seeman T, Pohl M, Misselwitz J, John U (2009) Angiotensin receptor blocker reduces proteinuria independently of blood pressure in children already treated with angiotensin-converting enzyme inhibitors. Kidney Blood Press Res 32:440–444
Lubrano R, Soscia F, Elli M, Ventriglia F, Raggi C, Travasso E, Scateni S, Di Maio V, Versacci P, Masciangelo R, Romero S (2006) Renal and cardiovascular effects of angiotensin-converting enzyme inhibitor plus angiotensin II receptor antagonist therapy in children with proteinuria. Pediatrics 118:e833–e838
Kaito H, Nozu K, Iijima K, Nakanishi K, Yoshiya K, Kanda K, Przybyslaw Krol R, Yoshikawa N, Matsuo M (2006) The effect of aldosterone blockade in patients with Alport syndrome. Pediatr Nephrol 21:1824–1829
Ku E, Campese VM (2009) Role of aldosterone in the progression of chronic kidney disease and potential use of aldosterone blockade in children. Pediatr Nephrol 24:2301–2307
The ONTARGET Investigators (2008) Telmisartan, ramipril, or both in patients at high risk for vascular events. N Eng J Med 358:1547–1559
Marrs JC (2010) Spironolactone management of resistant hypertension. Ann Pharmacother 44:1762–1769
Acknowledgements
The Alport Syndrome Treatments and Outcomes Registry (C.K., M.G., M.R., C.L.) receives support from private donors and the Alport Syndrome Foundation (www.alportsyndrome.org). The European Alport Registry is supported by the Association pour l’Information et la Recherche sur les Maladies Rénales Génétiques (AIRG) (to O.G.) and the KfH Foundation Preventive Medicine (Fritz-Scheler Stipendium of the German Society of Nephrology) (to O.G.). The EARLY PRO-TECT Alport trial is funded by the German Ministry of Education and Research (O.G.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kashtan, C.E., Ding, J., Gregory, M. et al. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol 28, 5–11 (2013). https://doi.org/10.1007/s00467-012-2138-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-012-2138-4