
Abstract
The paper presents possibilities and difficulties in nondestructive analysis of small multielement single crystals performed by means of X-ray spectrometry techniques: micro-X-ray fluorescence spectrometry (μ-XRF), energy-dispersive electron probe microanalysis (ED-EPMA), and X-ray photoelectron spectroscopy (XPS). The capability of the X-ray spectroscopy techniques in elemental analysis is demonstrated with the single crystals of selenide spinels of the general formula M x N y Cr z Se4 (M+2 and N+3 are, for example, Zn+2, V+3, Ga+3, Cd+2, In+3, and Sb+3). The results of the nondestructive analyses (μ-XRF, ED-EPMA, and XPS) are compared with those obtained by inductively coupled plasma optical emission spectrometry (ICP-OES) and wavelength-dispersive X-ray spectrometry (WDXRF) following sample digestion. The present study shows satisfactory agreement between the results of μ-XRF analysis performed using the standardless fundamental parameter method and the results obtained with the WDXRF and ICP-OES analyses. If the measured single crystal is precisely positioned, the difference between μ-XRF and wet analysis (WDXRF and ICP-OES) does not exceed 5% rel. The reliable results of ED-EPMA can be obtained only if the measured area is sufficiently large, i.e., of 200 × 300 μm. Even if this condition is fulfilled, the relative difference between the ED-EPMA and the wet analysis may reach 10% rel. In case of the XPS analysis, the accuracy of results depends on the proper preparation of the sample surface. It should be free of contamination that can be obtained by scraping in situ in ultrahigh vacuum. The ion etching, commonly used for cleaning the surface, leads to preferential sputtering; therefore, the reliable results cannot be obtained.

The paper presents possibilities and difficulties in nondestructive analysis of small multielement single crystals performed by means of X-ray spectrometry techniques: micro-XRF, ED-EPMA and XPS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chromium selenospinels are well-known ferromagnetic and antiferromagnetic semiconductors with unique electrical properties [1–4]. The general chemical formula of the compounds can be written as (A)[Cr2]Se4, where A2+ represents Cd2+, Zn2+, Cu2+, and Co2+ cations. The single crystals of the spinels are grown by a chemical vapour transport method with anhydrous chromium chloride as a transporting agent. This process leads to a growth of the single crystals of dimensions from approximately 0.5 to 5 mm (Fig. 1) and containing various amounts of the doped elements. Since chemical composition strongly influences electric and magnetic properties of these materials [1–3], detailed information on their stoichiometry is required. The chemical composition can be determined after sample digestion using, for example, inductively coupled plasma optical emission spectrometry (ICP-OES) [4] or wavelength-dispersive X-ray fluorescence spectrometry (WDXRF) [5, 6]. However, the nondestructive methods are much more desirable because investigation of the physical properties is usually carried out for the materials in crystalline state. X-ray fluorescence techniques are ideal tools for analysis of advanced materials of both bulk and thin layers [7–9]. Recently, the nondestructive analysis of monocrystals and polycrystals by using a laboratory-constructed energy-dispersive X-ray fluorescence spectrometry with monochromated primary radiation has been described [10].

The single crystals of Zn1 − x Cd x Cr2Se4 spinels
In the present paper, the application of other nondestructive X-ray spectroscopy methods for elemental analysis of the chromium selenospinels of the general formula M x N y CrzSe4 (where M2+ and N3+ are Zn2+, V3+, Ga3+, Cd2+, In3+, and Sb3+) is discussed. The synthesized crystals of various compositions were studied by micro-X-ray fluorescence spectrometry (μ-XRF), energy-dispersive electron probe microanalysis (ED-EPMA), and X-ray photoelectron spectroscopy (XPS). The results of the nondestructive techniques are compared with those obtained from ICP-OES and WDXRF, both requiring the digestion of specimen.
Experimental
μ-XRF
The single crystals of the spinels of dimensions from approximately 1 to 3 mm were analyzed without any previous treatment. The measurements were performed with the laboratory-constructed spectrometer described in details in [11]. The samples were excited by an X-ray beam from the air-cooled side-window Rh target of the X-ray tube of 125-μm thickness Be window and approximately 100-μm nominal focal spot size (XTF 5011/75, Oxford Instruments, USA). The X-ray tube was supplied with XLG high-voltage generator (Spellman, USA). The size of the analyzed area may be optimized with the tungsten pinhole collimators of the diameter of 100, 200, 400, 1,000, and 2,000 μm, ensuring the focal spot sizes of 169, 280, 581, 1,469, and 2,888 μm, respectively. The X-ray spectra emitted by the sample were collected by a thermoelectrically cooled Si-PIN detector (XR-100CR Amptek, Bedford, MA, USA) of 6-mm2 active area, 500-μm crystal thickness, and 12.5-μm Be window thickness. The resolution of the Si-PIN detector cooled to the temperature of approximately −55 °C was 145 eV at 5.9 keV. The detector was coupled to a multichannel analyzer (PX4 Amptek, Bedford, MA, USA). The sample position was adjusted by the X–Y stage and monitored with the CCD camera and two laser pointers.
EPMA
Microanalysis was performed with EDS analyzer (ISIS-300, Oxford Instruments) coupled to the scanning electron microscope (JSM-5410, JEOL). The accelerating voltage was 20 kV. The takeoff angle was 45°. The X-ray spectra were collected by liquid-nitrogen-cooled Si(Li) detector with energy resolution of 130 eV. The quantitative analysis was carried out using SEMQuant software (Oxford Instruments) and pure element standards from Agar Scientific.
XPS
The XPS spectra were obtained with monochromated Al Kα radiation using PHI 5700/660 Physical Electronic spectrometer. Its hemispherical analyzer ensures resolution of about 0.3 eV in kinetic energy of photoelectrons. Two different modes of purifying the sample from contaminations were applied and compared. In the first run of the experiment, the surface of the sample was scrapped in situ at 10−10-hPa vacuum. In the second run, the profiles were measured by sputtering the outer layer of the sample with contaminations at 1 kV with Ar+.
The following spectra were measured: (1) an overview of the binding energy in the range −2 to 1,400 eV; (2) the high-resolution spectrum of the valence band in the region −2 ÷ 20 eV; and (3) the core-level characteristic peaks for Se 3d, Cr, Cd, and Zn 2p 3/2 . The accurate chemical composition was determined from the core-level characteristic peaks. The background was subtracted using Tougaard’s approximation.
WDXRF
The single crystal was weighed on a microanalytical balance (the masses of analyzed samples were usually between 5 and 20 mg). Then the crystal was digested in a 20-mL baker in aqua regia (1.5 mL of concentrated HCl and 0.5 mL of concentrated HNO3). After 20 min of heating, the material became completely dissolved. The solution was transferred into a 10-mL volumetric flask and filled up to the mark with water. Next, 0.5 mL of the solution was pipetted onto the Whatman filter (21 mm in diameter) and dried under IR heater. The standard samples were prepared by pipetting appropriate solutions of analytes onto the Whatman filters. The prepared samples were analyzed using a wavelength-dispersive X-ray spectrometer with LiF(200) analyzing crystal and the silver target X-ray tube operating at 45 kV and 40 mA.
ICP-OES
The single crystals were digested in the same way as for the WDXRF measurements. The obtained solutions were measured using a sequential spectrometer with an excitation in the ICP plasma made by a SPECTRO Analytical Instruments (Germany) with the following parameters: frequency 27.12 MHz, power 1.1 kW, torch Ar/Ar/Ar (quartz, demountable), coolant gas 14.0 L min−1, auxiliary gas 0.5 L min−1, nebulizer gas 1.0 L min−1, nebulizer concentric Meinhard, nebulizer pressure 2.4 bar, spray chamber of glass, according to Scott, sample rate 1.0 mL min−1, observation height 11 mm, holographic grating 2,400 grooves per milliliter, dispersion of grating in first reciprocal order 0.55 nm mm−1, and wavelength range of monochromator 165–460 nm.
Results and discussion
The μ-XRF spectrometry is a valuable tool for the nondestructive multielemental analysis of small specimens. Moreover, this low-cost and non-time-consuming technique can successfully be applied in the screening analysis. Therefore, μ-XRF spectrometry is an ideal tool for the analysis of single crystals of selenide spinels synthesized by chemical vapour transport method. The chemical vapour transport process leads to a growth of a few dozen monocrystals of sizes from approximately 0.5 to 5 mm. Moreover, each monocrystal can be doped with various amounts of element to be interested.
The quantitative μ-XRF analysis was performed using the standardless fundamental parameter (FP) method based on the Sherman equation [12] and Pella et al. algorithm [13, 14] to calculate the X-ray tube spectrum. Our previously reported experiments [11] have shown that the applied pinhole collimators did not modify the spectral distribution of the X-ray tube that is important in FP methods, especially in the standardless analysis. Although μ-XRF spectrometry has many advantages, the accurate results for the single crystals can be obtained if several experimental requirements are met. To these belongs the crystal sample positioning in order to avoid an appearance of strong reflections of the Bragg diffraction. Figure 2 presents the X-ray spectra for Zn1 − x Cd x Cr2Se4 collected at various crystal positions. The high-intensity diffraction peaks can be observed if the incidence and takeoff angles are equal 45°. Two problems arise: the diffraction peaks can be misidentificated and first of all the calculated net intensities of fluorescent radiation can be overestimated or underestimated after deconvolution of the spectrum. If the analyzed crystal takes such an orientation that the incidence and takeoff angles are equal 40° and 50°, respectively, the diffraction peaks may be eliminated, as shown in Fig. 2. Thus, the question is whether the crystal orientation (and diffraction peaks) affects the quantitative analysis. The results of the μ-XRF measurements carried out for the crystal set in various positions are given in Table 1. They show that the diffraction peaks have influence on the quantitative analysis; especially, the Zn concentrations differ rather significantly. It should be emphasized that the results are normalized during the calculations; therefore, the determined element concentrations are strongly correlated.

X-ray spectra of Zn1 − x Cd x Cr2Se4 collected at various positions of crystal. The high-intensity diffraction peaks (D) can be observed if the incidence and takeoff angles are equal 45°
The diffraction peaks are not the only source of the potential error in the analysis. High-quality results from the standardless FP method can be obtained if the assumed geometry of measurements (incidence and takeoff angles) is assured. Fortunately, the synthesized crystals have well-formed flat faces that enables easy sample adjusting. Nevertheless, various positions of the sample may strongly influence the quantitative results of the μ-XRF analysis; therefore, this effect was checked in detail. Figure 3 shows the relationship between the intensity of fluorescent radiation of Cr and Se and the position of the Zn0.81Cd0.20Cr1.99Se4 crystal. Figure 3 presents also the Cr and Se concentration calculated by the standardless FP for each crystal position. It can be noticed that in a wide range of the sample positions the results are close to those obtained for the sample in the central position, even if the analyzed crystal is only partially irradiated by an X-ray beam. This can be explained by the fact that all elements in the crystal are quantified, and the results are normalized during the FP calculations. Table 2 presents the analysis for Zn0.81Cd0.20Cr1.99Se4 crystal of dimensions of 900 μm adjusted in the central position and ±600 μm off-center, where the specimen is only partially excited by the primary X-ray beam. In the off-center position, the fluorescent radiation is emitted not only from the front but also from the slanted faces of the crystal. Table 2 presents also the analysis performed with the 2,000-μm collimator, which gives the 2,888-μm focal spot size, thus larger than the crystal size. These results indicate that the best accuracy can be obtained if the crystal is precisely set in the central position.

The intensity of the fluorescent radiation of Cr and Se versus the position of the Zn0.81Cd0.20Cr1.99Se4 crystal. The crystal size is approximately 900 μm and the pinhole collimator 200 μm
Taking into account the effect of crystal position on the results of analysis, the precision under repeatability conditions was evaluated for several monocrystals. Table 3 gives exemplary analyses for Zn0.81Cd0.20Cr1.99Se4. The measurements were repeated for the same and for various positions of the crystal. The standard deviations (SD) calculated for various positions are two to four times higher than those calculated for the measurements carried out without moving the sample. In general, if the crystal is precisely positioned, the relative standard deviation (RSD) should not be higher than 2%.
The results of μ-XRF analysis obtained by using the standardless FP method were compared with those resulting from ICP-OES and WDXRF, requiring the sample digestion. For this purpose, the measurements were done for several crystals of ZnCr2Se4, CdCr2Se4, and CuCr2Se4 doped with V, Ga, In, and Cd [15–18]. The agreement between the results appears to be quite good, as the relative differences in the analyses do not exceed 5%. Figure 4 shows the relationships between the indium concentration in seven samples of Cu x In y Cr z Se4 determined by μ-XRF and WDXRF or ICP-OES. The mean relative difference between μ-XRF and WDXRF is approximately 3%, whereas between μ-XRF and ICP-OES it amounts approximately to 4%.

The concentration of indium determined by μ-XRF and WDXRF (ICP-OES) in seven crystals of Cu x In y Cr z Se4
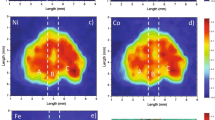
The higher values of SD for various positions of the crystal shown in Table 3 may result not only from the low precision in the crystal positioning but also from the inhomogeneous surface of the specimens. The surface inhomogeneity in our samples was investigated by ED-EPMA. To do so, five measurements were made in the same area (50 × 50 μm) of Zn0.81Cd0.20Cr1.99Se4 crystal and five other ones in various areas. The resulting analyses are collected in Table 4. They are rather repeatable for the data collected in the same area, whereas the measurements performed in various areas give dispersed results. The concentrations vary within the following ranges: 10.3–14.2%, 4.4–6.0%, 18.9–23.7, and 56.1–65.3% for Zn, Cd, Cr, and Se, respectively. This high dispersion can be explained by an inhomogeneous distribution of the ions in the spinel and also by a rough surface of the sample. Taking into account the inhomogeneity of the surface, the subsequent ED-EPMA measurements were carried out for the larger area of 200 × 300 μm. The exemplary results of μ-XRF and ED-EPMA analyses for ZnInCrSe and CdGaCrSe crystals, together with those from wet analyses (mean value of WDXRF and ICP-OES), are given in Table 5. In general, the relative differences between the nondestructive ED-EPMA and the wet analysis can reach 10%. Moreover, the very high errors should be expected for the elements at concentrations below 1% (m/m). It results from a nearness to the detection and quantity limits obtained by ED-EPMA. In case of low concentration of the doped elements in selenide spinel, the μ-XRF technique can successfully be applied. Detection limits for μ-XRF vary from 0.01% to 0.05% (m/m) depending on the element to be determined (experimental conditions: 400-μm pinhole collimator, 45 kV, 1,000 μA, 10-min counting time).
Considering further possibility of the nondestructive characterization of the selenide spinel crystals, the XPS method was also applied. XPS gives information only from the outer surface of the sample (up to approximately 10 nm); therefore, to evaluate the effect of the surface preparation on the results of analysis, two modes of measurements were checked. First, the chemical composition was analyzed based on the spectrum detected from the sample without previous surface cleaning (after about 1 h in vacuum, the first measurement was made and revealed at dirt surface, in atomic percents, 80.1% C, 16.4% O, 2.4% Se, 0.4% Cr, 0.4% Zn, 0.2% N, and 0.1% Cd). Next, the sample with a fresh surface obtained by scraping in situ in 10−10-hPa vacuum was measured (that time the composition was 62.4% Se, 22.1% Cr, 11.4% Zn, and 4.1% Cd, the contamination, i.e., C, O, and N, was less than 0.1%). Table 6 shows the results for Zn0.81Cd0.20Cr1.99Se4 crystal obtained with and without surface scraping; for comparison consistency in calculations for dirt surface, we omit contamination (C, O, and N). For comparison, the results of the μ-XRF, ED-EPMA, and wet analysis are also given. It turned out that the reliable chemical composition can be obtained only if the measurement is made for the fresh surface prepared by scraping in situ in 10−10-hPa vacuum. The accuracy of 10% is typically quoted for atomic concentrations routinely determined from the XPS experiments. However, the quantitative chemical analysis relies on several factors; therefore, standard assumptions may result in enormous errors in quantification [19, 20]. Also, the contamination of the surface is a source of serious errors. The best method in obtaining a free of contamination fresh surface is cleaving or scraping the crystal in situ in ultrahigh vacuum. The ion etching, commonly used for surface cleaning, may cause preferential sputtering in multielemental samples that leads to a change in their chemical composition. Besides, the investigations show that not only the preferential sputtering takes place (Fig. 5) but also some amount of contamination still remains. Moreover, in effect of the etching, the crystal surface may be deteriorated. The case is illustrated in Fig. 6, where the splitting of the 2p 3/2 peak disappears after the sputtering. Moreover, a small broadening of the XPS bands is observed. The 2p 3/2 peak splitting is typical for Cr3+ and can be observed only in the high-resolution measurements of surface of good-quality single crystals. The nature of the splitting is described in the literature [3].

Depth profiling analysis for the sample (ZnCdCrSe) without previous surface cleaning (a) and after scrapping the surface (b)

Chromium 2p core levels for the contamination-free crystal fresh surface obtained by scrapping in ultrahigh vacuum and after sputtering. The surface deterioration by sputtering reveals as a disappearance of the 2p 3/2 peak splitting
Conclusion
The present study shows satisfactory agreement between the results of the standardless μ-XRF analysis made by using the fundamental parameters method and the results obtained with the techniques requiring sample digestion (WDXRF and ICP-OES analysis using reference samples). The best accuracy can be obtained if the measured single crystal is precisely positioned. Then the difference between μ-XRF and wet analysis does not exceed 5% rel. The reliable results of ED-EPMA can be obtained only if the measured area is sufficiently large, i.e., of 200 × 300 μm. Even if this condition is fulfilled, the relative difference between the nondestructive ED-EPMA techniques and the wet analysis may reach 10%. Moreover, the high errors should be expected in the determination of elements at the concentrations below 1% (m/m). In case of the XPS analysis, the accuracy of results depends on the proper preparation of the sample surface. It should be free of contamination that can be obtained by scraping in situ in ultrahigh vacuum. The ion etching, commonly used for cleaning the surface, leads to preferential sputtering; therefore, the reliable results cannot be obtained.
References
Rudolf T, Kant Ch, Mayr F, Hemberger J, Tsurkan V, Loidl A (2007) Phys Rev B 76:174307–174310
Groń T, Mazur S, Duda H, Krok-Kowalski J, Malicka E (2007) Phys B 391:371–379
Malicka E, Waśkowska A, Heimann J, Mydlarz T, Sitko R, Kaczorowski D (2008) J Solid State Chem 181:1970–1976
Krok-Kowalski J, Warczewski J, Duda H, Gusin P, Krajewski K, Śliwińska T, Pacyna A, Mydlarz T, Malicka E, Kita A (2007) J Alloys Compd 430:39–42
Jurczyk J, Sitko R, Zawisza B, Buhl F, Malicka E (1999) Microchim Acta 132:41–47
Jurczyk J, Buhl F, Wilczek I (1993) Chem Anal (Warsaw) 38:519–526
Queralt I, Ibañez J, Marguí E, Pujol J (2010) Spectrochim Acta B 65:583–586
Baake O, Hoffmann PS, Flege S, Ortner HM, Gottschalk S, Berky W, Balogh AG, Ensinger W, Beckhoff B, Kolbe M, Gerlach M, Pollakowski B, Weser J, Ulm G, Haschke M, Blokhina E, Peter M, Porta D, Heck M (2009) Anal Bioanal Chem 393:623–634
Mino L, Agostino A, Codato S, Lamberti C (2010) J Anal At Spectrom 25:831–836
Sitko R, Zawisza B, Malicka E (2008) Spectrochim Acta B 63:1303–1308
Sitko R, Zawisza B, Malicka E (2009) Spectrochim Acta B 64:436–441
Sherman J (1955) Spectrochim Acta 7:283–306
Pella PA, Feng L, Small JA (1985) X-Ray Spectrom 14:125–135
Pella PA, Feng L, Small JA (1991) X-Ray Spectrom 20:109–110
Waśkowska A, Gerward L, Staun Olsen J, Morgenroth W, Malicka E, Skrzypek D (2008) J Phys Condens Mater 20:425209
Malicka E, Groń T, Pacyna AW, Maciążek E, Duda H, Pawełczyk M, Zawisza B, Sitko R (2009) J Alloys Compd 480:67–69
Malicka E, Waśkowska A, Skrzypek D, Sitko R, Kaczorowski D (2009) J Solid State Chem 182:3149–3154
Malicka E, Groń T, Skrzypek D, Pacyna AW, Badurski D, Waśkowska A, Mazur S, Sitko R (2010) Philos Mag 90:1525–1541
Tougaard S (1996) Appl Surf Sci 100/101:1–10
Tougaard S (2010) J Electron Spectrosc 178–179:128–153
Acknowledgments
The work was supported by the Ministry of Science and Higher Education by grant no. N N204 145938.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Focus on Analytical Science in Poland (VIII Polish Conference on Analytical Chemistry) with Guest Editor Pawel Koscielniak
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Malicka, E., Sitko, R., Zawisza, B. et al. Nondestructive analysis of single crystals of selenide spinels by X-ray spectrometry techniques. Anal Bioanal Chem 399, 3285–3292 (2011). https://doi.org/10.1007/s00216-010-4240-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4240-4