
Abstract
The Grasvally Norite–Pyroxenite–Anorthosite (GNPA) member within the northern limb of the Bushveld Complex is a mineralized, layered package of mafic cumulates developed to the south of the town of Mokopane, at a similar stratigraphic position to the Platreef. The concentration of platinum-group elements (PGE) in base metal sulfides (BMS) has been determined by laser ablation inductively coupled plasma–mass spectrometry. These data, coupled with whole-rock PGE concentrations and a detailed account of the platinum-group mineralogy (PGM), provide an insight into the distribution of PGE and chalcophile elements within the GNPA member, during both primary magmatic and secondary hydrothermal alteration processes. Within the most unaltered sulfides (containing pyrrhotite, pentlandite, and chalcopyrite only), the majority of IPGE, Rh, and some Pd occur in solid solution within pyrrhotite and pentlandite, with an associated Pt–As and Pd–Bi–Te dominated PGM assemblage. These observations in conjunction with the presence of good correlations between all bulk PGE and base metals throughout the GNPA member indicate the presence and subsequent fractionation of a single PGE-rich sulfide liquid, which has not been significantly altered. In places, the primary sulfides have been replaced to varying degrees by a low-temperature assemblage of pyrite, millerite, and chalcopyrite. These sulfides are associated with a PGM assemblage characterized by the presence of Pd antimonides and Pd arsenides, which are indicative of hydrothermal assemblages. The presence of appreciable quantities of IPGE, Pd and Rh within pyrite, and, to a lesser, extent millerite suggests these phases directly inherited PGE contents from the pyrrhotite and pentlandite that they replaced. The replacement of both the sulfides and PGM occurred in situ, thus preserving the originally strong spatial association between PGM and BMS, but altering the mineralogy. Precious metal geochemistry indicates that fluid redistribution of PGE is minimal with only Pd, Au, and Cu being partially remobilized and decoupled from BMS. This is also indicated by the lower concentrations of Pd evident in both pyrite and millerite compared with the pentlandite being replaced. The observations that the GNPA member was mineralized prior to intrusion of the Main Zone and that there was no local footwall control over the development of sulfide mineralization are inconsistent with genetic models involving the in situ development of a sulfide liquid through either depletion of an overlying magma column or in situ contamination of crustal S. We therefore believe that our observations are more compatible with a multistage emplacement model, where preformed PGE-rich sulfides were emplaced into the GNPA member. Such a model explains the development and distribution of a single sulfide liquid throughout the entire 400–800 m thick succession. It is therefore envisaged that the GNPA member formed in a similar manner to its nearest analogue the Platreef. Notable differences however in PGE tenors indicate that the ore-forming process may have differed slightly within the staging chambers that supplied the Platreef and GNPA member.
Similar content being viewed by others
Introduction
The Bushveld Complex, South Africa, is the world’s largest repository of platinum-group elements (PGE). The Complex comprises a package of layered ultramafic and mafic cumulates named the Rustenburg Layered Suite, present in five geographically distinct limbs (Fig. 1) and divided into five stratigraphic units. The PGE reserves are present within three main deposits: the UG2 chromitite, the Merensky Reef, and the Platreef. Within the eastern and western limbs of the intrusion, PGE mineralization is confined to thin, stratiform layers in association with sulfides or chromitites. The most important of these, the Merensky Reef and UG2 chromitite are located towards the top of the most economically important unit: the Critical Zone. Within the northern limb, Platreef mineralization is present within a 10–400 m thick basal unit, intruded as a series of sills (Kinnaird 2005) that rests directly on Palaeoproterozoic sediments and Archaean gneisses and granites and is overlain by Main Zone gabbronorites. Widespread contamination of the Platreef magma through assimilation of differing floor rocks along its strike length largely accounts for the complexity of the deposit, which formed through the interaction of magmatic, metasomatic, and hydrothermal processes (e.g., Harris and Chaumba 2001; Armitage et al. 2002; Manyeruke 2003; Hutchinson and Kinnaird 2005; Kinnaird 2005; Kinnaird et al. 2005; Manyeruke et al. 2005; Sharman-Harris et al. 2005; Holwell and McDonald 2006, 2007; Holwell et al. 2006, 2007, 2011; Hutchinson and McDonald 2008; McDonald et al. 2009; Sharman et al. 2013).

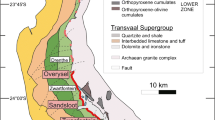
The Platreef (sensu stricto) is present only north of the Ysterberg–Planknek Fault (Kinnaird and McDonald 2005; Fig. 1), and represents a package of texturally heterogeneous and variably altered pyroxenitic lithologies, which is irregularly mineralized with sulfide associated PGE, Ni, and Cu (e.g., Armitage et al. 2002; Kinnaird 2005; Holwell et al. 2006; Holwell and McDonald 2006; Hutchinson and Kinnaird 2005; Hutchinson and McDonald 2008; Manyeruke et al. 2005; McDonald and Holwell 2011). To the south of Ysterberg–Planknek Fault, a distinct layered package of PGE-bearing mafic cumulates termed the Grasvally Norite–Pyroxenite–Anorthosite (GNPA) member is developed (Hulbert 1983). The GNPA member is present at a similar stratigraphic position to the Platreef, being overlain by Main Zone gabbronorites and resting directly on both Lower Zone ultramafic/mafic cumulates and the Magaliesberg Quartzite Formation from the Palaeoproterozoic Transvaal Supergroup. In previous studies, the GNPA member has been assumed to correlate with the Platreef (e.g., von Gruenewaldt et al. 1989; van der Merwe 1976, 2008; Maier et al. 2008) and possibly with the Critical Zone of the eastern and western limbs (von Gruenewaldt et al. 1989; van der Merwe 2008; Dunnett et al. 2012; Grobler et al. 2012). Since McDonald et al. (2005a, b) challenged this proposed correlation the relationship of the GNPA member with the Platreef has been under review (see also McDonald and Holwell 2011).
A recent study by Smith et al. (2011) concentrating on the sulfide mineralogy concluded that the presence of two distinct sulfide assemblages reflects the involvement of both magmatic sulfide fractionation processes and low temperature fluid alteration (<230 °C) in the development of sulfide mineralization within the GNPA member. At present, the factors involved in ore genesis with regards to the timing of S saturation relative to emplacement, the role of sulfides in concentrating PGE, and the effect of postmagmatic hydrothermal fluids are not well constrained.
Typically, contact-style PGE–Ni–Cu mineralization similar to that present within the GNPA member and Platreef is often attributed to the development of an immiscible sulfide liquid through in situ contamination by assimilation of crustal S (e.g., Duluth Complex; Mainwaring and Naldrett 1977; Ripley 1981; Ripley et al. 1986 and the Basal Series of the Stillwater Complex; Lambert et al. 1994; Lee 1996; McCallum 1996). Within the Platreef, it is now accepted that early contamination at depth induced S saturation, with localized contamination acting only as an ore-modifying process (Holwell et al. 2007; McDonald and Holwell 2007; Penniston-Dorland et al. 2008; Ihlenfeld and Keays 2011). The Platreef is also an example where hydrothermal fluids and contamination have had a significant influence over the resulting mineralogy and distribution of PGE (Hutchinson and Kinnaird 2005; Kinnaird 2005; Holwell and McDonald 2006, 2007; Holwell et al. 2006; Hutchinson and McDonald 2008). The complexity of the Platreef highlights that, in order to gain a full understanding of the ore genesis of any PGE–Ni–Cu deposit, it is critical to assess in detail the effects of magmatic, contamination, and hydrothermal processes. Considering the lesser known GNPA member, the presence of primary and secondary sulfide assemblages strongly suggests that both magmatic and hydrothermal processes are the major factors involved in the generation and distribution of PGE and BMS mineralization. In this paper, we investigate the precise distribution and mineralogy of PGE within the GNPA member to establish the role played by sulfide liquid in the concentration of PGE and the effects of postmagmatic fluids on the mineralogy and distribution of PGE. We also explore the processes involved in ore genesis, with particular interest on constraining the timing of S saturation relative to emplacement, by comparing the GNPA mineralization with its nearest analogue the Platreef and more widely with the Merensky Reef (van der Merwe 1976, 1978, 2008; Hulbert 1983; Maier et al. 2008).
Regional geological setting
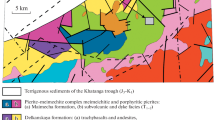
The 2.06 Ga Bushveld Complex covers an area of about 65,000 km2 and is the world’s largest layered igneous intrusion. The complex comprises five limbs (Fig. 1): the near symmetrical western and eastern limbs; a southern limb, partially hidden by younger sediments; a heavily eroded far western limb; and a northern limb (Eales and Cawthorn 1996). The Bushveld Magmatic Province as a whole comprises the felsic volcanics of the Rooiberg Group (Twist 1985; Buchanan et al. 2002), the mafic–ultramafic layered rocks of the Rustenburg Layered Suite, the Rashoop Granophyre Suite (Walraven 1985), the Lebowa Granite Suite (Walraven and Hattingh 1993), and a set of marginal pre- and syn-Bushveld sills (Cawthorn et al. 1981) (Fig. 1). The Rustenburg Layered Suite consists of a 7–8 km thick layered package, which is conventionally subdivided into five major stratigraphic zones: Marginal Zone norites, Lower Zone pyroxenites and harzburgites, Critical Zone chromitite–pyroxenite–norite cyclic units, Main Zone homogeneous gabbronorites and Upper Zone anorthosites, ferrogabbros, and magnetites. In the northern limb, the mafic succession deviates from the conventional Bushveld stratigraphy. The Platreef/GNPA member may represent the stratigraphic equivalent to the Critical Zone of the eastern and western limbs. Furthermore, Lower Zone cumulates are unusually thick (800–1,600 m), compared to that in the other limbs (van der Merwe 1976).
The GNPA member, present south of the Ysterberg–Planknek Fault, comprises vari-textured gabbronorites, norties, anorthosites, pyroxenites, and a PGE-bearing chromitite. The 400–800 m thick succession differs from the pyroxenitic Platreef in that it can be subdivided into three distinct stratigraphic units (Fig. 2; de Klerk 2005): the Lower Mafic Unit (LMF), the Lower Gabbronorite Unit (LGN), and the Mottled Anorthosite Unit (MANO). The LMF is distinguished from the homogeneous gabbronorites of the LGN by an increase in melanocratic lithologies, the development of two chromitite layers, and elevated bulk Cr values. The MANO is recognized by a substantial increase in plagioclase cumulates and the development of lithologies such as mottled and spotted anorthosites (Hulbert 1983; Smith et al. 2011). To the east of the N–S trending Grasvally Fault (Fig. 2), the GNPA member forms a plunging syncline directly overlying interbedded quartzites and shales of the Magaliesberg Quartzite Formation (van der Merwe 2008). West of the Grasvally Fault Lower Zone cumulates underlie the GNPA member (Fig. 2). Northwards the base of the Rustenburg Layered Suite, represented by the Platreef, progressively transgresses downwards through interbedded quartzites and shales of the Magaliesberg Quartzite Formation, quartzites and shales of the Timeball Hill Formation, shales of the Duitschland Formation, the Penge banded iron formation, the Malmani Subgroup dolomites to rest on Archaean basement granites, and gneisses in the far north (e.g., Sharman-Harris et al. 2005; Holwell and McDonald 2006; van der Merwe 2008; Fig. 1).

Detailed map of the GNPA member in the Rooipoort–Grasvally region accompanied by stratigraphic column. Locality of boreholes sampled is also shown. Adapted from Maier et al. (2008)
Within the GNPA member, PGE and BMS mineralization is not lithologically bounded, with wide but irregular zones developed throughout the LMF and MANO units (Maier et al. 2008). Mineralization associated with a chromite layer positioned within the basal LMF unit represents the only traceable horizon throughout the GNPA member in the Rooipoort and Grasvally region.
Samples and methods
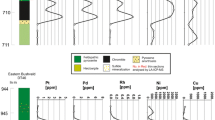
Samples of quarter core have been obtained from eight boreholes drilled by Falconbridge Ltd. and Caledonia Mining on the farms Rooipoort, Grasvally, and Moorddrift (Fig. 2) where the GNPA member overlies Lower Zone harzburgites and the Magaliesberg Quartzite Formation. A stratigraphic log of borehole RP04.23 provides a representative section of the entire GNPA member (Fig. 3), with the log of borehole RP05.45 showing differences in the succession where underlain by floor quartzites. These logs also highlight the position of mineralized zones identified by the presence of visible BMS and indications of PGE grades.

Detailed stratigraphic logs of boreholes RP04.23 and RP05.45 of the Lower Mafic (LMF), Lower Gabbronorite (LGN), and Mottled Anorthosite (MANO) Units, highlighting zones of visible sulfide mineralization and indication of PGE grades. Lithological abbreviations: SA spotted anorthosite, MA mottled anorthosite, GBN gabbronorite, PYX pyroxenite, FPX feldspathic pyroxenite
In total, 36 polished thin sections were analyzed for PGMs at the University of Leicester using a Hitachi S-3600N environmental scanning electron microscope, coupled to an Oxford Instruments INCA 350 energy dispersive X-ray analysis system.
Bulk concentrations of PGE and Au were determined at Cardiff University by Ni sulfide fire assay with Te coprecipitation followed by inductively coupled plasma–mass spectrometry (ICP-MS) procedure, following the methodology described by Huber et al. (2001) and McDonald and Viljoen (2006). The proportions required for fusion of a 15 g sample were 6 g of NaCO3, 12 g of borax, 0.9 g of sulfur, 1.08 g of carbonyl-purified Ni, and 1 g of silica. The flux for samples containing >50 % chromite contained 5 g of sample, 12 g of NaCO3, 24 g of Li Tetraborate, 0.9 g of sulfur, 1.08 g of carbonyl-purified Ni, 10 g of silica, and 2.5 g of NaOH. All samples were fired in fire-clay crucibles at 1,050 °C for 90 min. The sulfide buttons were dissolved in concentrated HCl. Noble metals that had entered the solution were coprecipitated with Te using SnCl2 as a reductant. Finally, soluble PGE chloro-complex solutions were spiked with Tl, which acts as an internal standard, enabling instrumental drift to be monitored during ICP-MS.
Whole-rock sulfur concentrations were determined by standard combustion iodometric procedures using a Laboratory Equipment Company titrator at the University of Leicester. Depending on the sulfide content between 0.05 and 0.2 g of sample was combusted for each titration. The rerunning of blanks, standards, and samples in triplicate ensured consistent results were obtained. The standard deviations of weight percent of sulfur ranged from 0.0005 to 0.2, indicating a high level of precision.
Sulfide analyses were carried out using a New Wave Research UP213 UV laser system coupled to a Thermo X Series 2 ICP-MS. The relative abundances of PGE and other elements were recorded in time-resolved analyses mode (time slices of 250 ms) as the laser beam followed a line designed to sample different sulfide or oxide phases. The beam diameter employed was 30 μm, with a frequency of 10 Hz and a power of ∼6 J cm−2. The sample was moved at 6 μm s−1 relative to the laser along a predetermined line pattern. Ablations were carried out under helium (flow, ∼0.7 L min−1), and the resulting vapor combined with argon (flow rate, 0.65–0.75 L min−1) before delivery to the ICP-MS. Acquisitions lasted between 80 and 400 s, including a 20 s gas blank prior to the start of the analysis and a 10 s washout at the end. Signals within the time spectra that could be attributed to PGM included in the sulfides were not selected for integration so the data reflect concentrations in the sulfide minerals alone. Sulfur concentrations were measured prior to laser ablation (LA)-ICP-MS using the electron microprobe at the University of Leicester and 33S was used as internal standard as some sulfides did not contain Fe. Subtraction of gas blanks and internal standard corrections were performed using Thermo Plasmalab software.
Calibration was performed using a series of 5 synthetic Ni–Fe–S standards prepared from quenched sulfides. The standards incorporate S, Ni, Fe, and Cu as major elements and Co, Zn, As, Se, Ru, Rh, Pd, Ag, Cd, Sb, Te, Re, Os, Ir, Pt, Au, and Bi as trace elements and the compositions of the five standards are given in Prichard et al. (2013). The standards produce five point calibration curves for S, Ni, and Fe and three point calibration curves for PGE, Ag, Cd, Re, Au, and semimetals. Standards 1–3 produce three point calibration curves for Cu, Co, and Zn and reliable matrix-matched corrections for argide species (59Co40Ar, 61Ni40Ar, 63Cu40Ar, 65Cu40Ar, and 66Zn40Ar) that interfere with 99Ru, 101Ru, 103Rh, 105Pd, and 106Pd. Corrections for 106Cd on 106Pd and 108Cd on 108Pd were determined using Cd-bearing Standard 1, but Cd concentrations in the sulfides were <10 ppm, producing only very small corrections in most unknowns. Argide and isobaric-corrected data are indicated by asterisks beside 101Ru, 103Rh, 105Pd, 106Pd, and 108Pd in the relevant tables. Where independent corrections have been applied to different isotopes of the same element (e.g., 66Zn40Ar and 106Cd on 106Pd and 108Cd on 108Pd), the independently corrected values typically vary by <20 % (and commonly <5 %) indicating that the corrections are robust. The accuracy of the LA-ICP-MS procedure for PGE was checked by analysis of the Laflamme-Po724 standard run as an unknown against the Cardiff sulfide standards at the start and end of each day.
Platinum-group mineralogy
Thirty-six polished thin sections from the quartzite floor rocks, LMF (including chromitites) and MANO from boreholes RP04.23, RP05.45, MD03.1, RP04.21, and RP05.37 were examined for PGM. More than 800 individual PGM grains have been identified and are listed in Table 1. Each individual grain has been classified by its composition, size, rock type, and associated BMS assemblage (primary or secondary). The relative proportions of the various PGM are based on an estimation of area (and by inference, volume) of each grain. This was calculated using the short- and long-axes of PGM, measured on the SEM. To prevent biases, we present all data on PGM assemblages in percentage of total area of all PGM, which reflects more accurately the relative proportions of each PGM type within an assemblage. Each occurrence was also classified by its association: enclosed in sulfide, attached to sulfide, enclosed by silicates, or attached/enclosed within chromite or oxide (Fig. 4).

Backscattered electron photomicrographs of PGM found within the GNPA member. a, b Pt/Pd-bearing phases found attached and enclosed within pentlandite (pn), chalcopyrite (cpy), and pyrite (py). c Cluster of PGM enclosed fully in pyrite and millerite (mil). d, e PGM residing in quartz and secondary silicates (e.g., actinolite, tremolite and chlorite), in close proximity to pyrrhotite (po) dominated sulfide blebs. f PGM within a chromitite showing association to sulfide over chromite (cr)
PGM assemblages
Within the GNPA member, the PGE mineralogy is dominated by Pt–As and Pd–Bi-Te-bearing PGM. Platinum- and Pd-bearing phases constitute 53 and 35 % (by area), respectively, of all PGM classified. The identified PGM have been grouped into a total of 11 types (see Table 2). The five most abundant by area are (Table 2) (1) Pt arsenides (50 %), (2) Pd bismuthotellurides (15 %), (3) Pt–Pd tellurides (14 %), (4) Pd antimonides (10 %), and (5) Au–Ag minerals (8 %). No PGM carriers of Os or Ir were observed within this study. In addition, throughout the Rooipoort, Grasvally, and Moorddrift area, PGE sulfides in the form of laurite, cooperate, and braggite along with Pt–Pd–Fe alloys are rare forming <0.05 % of the total assemblage by area (Tables 1 and 2).
No noticeable differences exist between the PGM assemblages developed in the MANO unit, LMF unit, and footwall rocks. Mineralization within the latter is interpreted to result from infiltration of the sulfide liquid into the footwall. With the exception of the chromitites (Tables 1 and 2), the PGE mineralogy also does not vary considerably with lithology. The proportions of PGM types do however differ quite significantly between primary and secondary sulfides, indicating sulfide assemblage is the controlling factor on PGM assemblage (Tables 1 and 2; Smith et al. 2011). Thus, we regard sulfide mineral assemblage as the primary control on differing PGE mineralogies, and the following sections are structured accordingly to this distinction.
Non chromitiferous rocks and quartzites
Primary sulfide assemblages
The PGE mineralogy associated with the primary pyrrhotite–chalcopyrite–pentlandite sulfide assemblage is overwhelmingly dominated by Pt arsenides, specifically sperrylite (PtAs2), which forms around 70 % of the total PGM assemblage (Table 2). Sperrylite, however, represents only eight out of the 273 grains identified within primary sulfide-bearing samples (Table 1); thus, a significant proportion of the area is contributed by a single grain with the dimensions 127 μm × 65 μm. Therefore, the apparent dominance of sperrylite should be treated with caution on consideration of this potential nugget effect. The remaining assemblage consists primarily of the Pd bismuthotelluride michenerite (13 %) and Pt–Pd tellurides (9 %) with Au and Ag minerals such as electrum and hessite constituting only 2 %. It is important to highlight the rather low abundance of Sb-bearing PGM (3.6 %) within the primary sulfide-bearing samples (Table 2).
The PGM consistently appear to be closely associated with sulfide (e.g., Fig. 4) with 48 % of PGM residing fully enclosed within sulfides (primarily pyrrhotite and pentlandite) or existing along the sulfide margins (Fig. 4a, b; Table 3). Although a significant proportion (51 %) of PGM occur as satellite grains within secondary silicates, they remain spatially in close association with BMS (e.g., Fig. 4d, e).
Secondary sulfide assemblages
Where primary sulfides are replaced to varying extents by pyrite and millerite, the types and proportions of PGM vary from those discussed above (Tables 1 and 2). Although sperrylite continues to dominate the assemblage, the proportion of Pt-bearing PGM is notably lower at around 44 %. The most significant difference however is the increase in the proportion of Pd antimonides (12 %) and the appearance of Pd arsenides, such as palladoarsenite (Pd2As; Tables 1 and 2). The rest of the assemblage is, in general, comparable to that described above, comprising Au and Ag minerals, Pt–Pd tellurides (each accounting for around 14 %), and Pd bismuthotellurides (12 %). More obscure phases identified that are unique to the secondary sulfides include sopcheite (Ag4Pd3Te4), maslovite (PtBiTe), isomertieite (Pd11Sb2As2), and temagamite (Pd3HgTe3; Table 1; Fig. 4c). The latter is relatively rare within the northern limb, reported only once within the Platreef, at Tweefontein (McCutcheon and Kinnaird 2011). The associations of PGM are similar to those in the primary assemblages, with a strong relationship remaining between PGM and BMS with 45 % of the PGM assemblage residing in alteration silicates (mainly chlorite, tremolite, and actinolite) or quartz surrounding/replacing the sulfide bleb (Table 3; Fig. 4d, e). The rest of the assemblage (>50 %) mainly exists in direct association with the sulfides, occurring both along the margins of and fully enclosed within sulfide minerals (Fig. 4c; Table 3). Pyrite and millerite are the dominant hosts of PGM inclusions, with few occurring within the relicts of primary pyrrhotite, pentlandite, and chalcopyrite.
Chromitiferous rocks
The PGM assemblage of the chromitites is broadly comparable to that of the chromite-poor rocks with Pt–Pd arsenides, Pd bismuthotellruides, and Pt–Pd tellurides dominating. However, minor but highly significant differences do exist, including the appearance of Ru sulfide and the higher abundance of PGE sulfarsenides (Tables 1 and 2). Within the chromitites, although the presence of chromite exerts a minor control over the platinum-group mineralogy, it appears to be principally controlled by the sulfide assemblage developed along with the spinels (Table 2).
Primary sulfide assemblages
Although the sample size is significantly lower than observed within the nonchromitiferous rocks of the GNPA member, this study has still managed to reveal that, while the PGM assemblage is dominated by Pd bismuthotellurides (42 %) and Pt arsenides (27 %), it is notably distinct due to the presence of laurite (RuS2) and the greater proportion of PGE sulfarsenides, principally hollingworthite (RhAsS; Tables 1 and 2; Fig. 4f).
The PGM exhibit a strong preference to BMS rather than chromite (Table 3). Overall, 44 % occur in direct association with sulfides, present either fully enclosed within pentlandite or along margins of sulfides (Fig. 4f). A comparable percentage of PGM were found within alteration silicates, as satellite grains surrounding BMS. The PGM are rarely found in association with the chromite grains with only one grain (11 % of the assemblage) attached to chromite (Table 3). No PGM were found included within chromite grains.
Secondary sulfide assemblages
Chromitites with secondary textured sulfides have a PGM assemblage that is near comparable to other secondary sulfide-bearing rocks of the GNPA member (Table 2). The assemblage consists primarily of Pd bismuthotellurides (30 %), Pt arsenides (25 %), and Pt–Pd tellurides (24 %), and shows considerably diversity in PGM type (Table 2). Further similarities include the rather high abundance of Pd antimonides, which account for 16 % of the assemblage. The platinum-group mineralogy within the GNPA member appears to therefore be more strongly controlled and/or related to the development of secondary sulfides than the presence of chromite.
As observed within the chromitites hosting primary textured sulfides, the PGM show greater preference to BMS than chromite (Table 3 and Fig. 4f). A combined total of 58 % were found in direct association with sulfides, thus situated fully enclosed or along the sulfide–silicate boundary (Table 3). Those enclosed in sulfide were generally hosted by pyrite and millerite. A high proportion of the PGM (40 %) also reside within secondary silicates surrounding BMS. A close association between PGM and chromite is not observed with only one grain found attached to chromite (Table 3). No PGMs were found as inclusions within the chromite.
PGE and base metal geochemistry
Throughout the GNPA member, PGE and BMS mineralization is typically confined to irregular zones that range in thickness from a few meters to ≥50 m (Fig. 3), hosted by a range of rock types, including chromitites. Mineralization also extends for several meters into the underlying quartzites. Whole-rock concentrations of S, Ni, Cu, and PGE on Grasvally, Rooipoort, and Moorddrift are listed in Table 4. The GNPA member is Pd dominant, with Pt/Pd ratios of the non chromitiferous rocks ranging between 0.1 to1.7 (mean, 0.5) and Ni/Cu ratios of the mineralized samples (defined as samples with Cu >400 ppm; Ni >1,000 ppm) between 0.4 and 4 (mean 1.6). The ore-body is characterized by variable PGE grades from sub-economic (<0.1 ppm 3PGE + Au) to high grade (>4 ppm), with the latter associated primarily with the chromitites (Table 4). In general, high Cu (>400 ppm) and Ni (>1,000 ppm) concentrations broadly correspond to high S contents and are also indicative of elevated PGE grades. This correlation is however less well defined within those samples hosting secondary sulfides.
In order to gain an insight into the controlling effects of magmatic and hydrothermal processes on the distribution of PGE, it is important to assess in detail the relationship between PGE and BMS within those samples containing secondary sulfides (Fig. 5). In a similar manner to the approach used for the PGM assemblages, we address these relationships with relation to the primary and secondary sulfide assemblages.

Binary variation diagrams plotting bulk rock: a Pt vs Pd, b Rh vs Pd, c Rh vs Pt, d Ir vs Pt, e Ir vs Pd, f Ru vs Ir, g Ir vs Au, h Pd vs Au, i Cu vs Ni, j Cu vs S, k Cu vs Pt, and l Cu vs Au
Non chromitiferous rocks and quartzites
Primary sulfide assemblages
Selected PGE are plotted against each other in Fig. 5 where a high degree of correlation is evident between the PGE in primary sulfide-bearing samples (Fig. 5a–f). The base metals (Cu and Ni) also correlate well with each other and with the PGE (Fig. 5i). Good correlations are also observed between PGE, Ni, and Cu with S (Fig. 5j). Gold appears to also be strongly associated with PGE in the most unaltered GNPA member rocks (Fig. 5g, h). The Pt/Pd ratio of primary sulfides is well constrained ranging between 0.2 and 0.5 (mean, 0.3; Table 4). The Ni/Cu ratio ranges from 0.4 to 3.7, with a mean of 1.4. The Pd/Ir and Rh/Ir ratios are consistent both between units and sulfide assemblages (Table 4; Fig. 6). The Pd/Ir ratio is rather variable ranging typically between 30 and 400. The Rh/Ir ratio varies between 2 and 8, with a mean of 3.8.

Ratios with depth through borehole RP04.23 for primary and secondary sulfide-bearing rocks for a Pt/Pd, b Pd/Ir and Rh/Ir, and c Ni/Cu. MA mottled anorthosite, FPYX feldspathic pyroxenite, GBN gabbronorite, PYX pyroxenite
Secondary sulfide assemblages
Strong positive correlations remain between the IPGEs, which is especially apparent between Ir and Ru (Fig. 5f). Both Rh and Pt correlate fairly well with the IPGEs and with each other (Fig. 5c and d), with only a slight scatter observed in the data set. In comparison, Pd, Au, and, to a lesser extent, Cu exhibit noticeably poorer correlations with the other PGE and especially with those that are considered immobile under most conditions (Pt and Ir; Fig. 5a, b, e, g, and k; Keays et al. 1982; Wood 2002). Interestingly, with the exception of two anomalous samples, Pd and Au continue to be strongly correlated with each other, even where alteration has occurred (Fig. 5h). Copper does not show any relationship with Au (Fig. 5l). Broad correlations are evident between Pd and Pt and also Rh, (Fig. 5a, b) although not as confined as those observed within the primary sulfide-bearing samples. No relationship is preserved between Pd and Ir (Fig. 5e). The base metals and PGE do not continue to be closely associated with S, with a much broader relationship evident (Fig. 5j). Copper and Ni do however remain generally well correlated with each other (Fig. 5i). Both the Pt/Pd ratio (mean of 0.6) and Ni/Cu ratio (mean of 1.8) are slightly elevated within the secondary sulfides in comparison to those samples hosting primary sulfides. All of these observations are consistent with the preferential remobilization of Pd and Au over the rather more immobile Pt, IPGE, and Rh (Wood 2002), by late-stage hydrothermal fluids.
Chromitiferous rocks
It is noticeable from Fig. 5 that the chromitites, which are both primary and secondary sulfide-bearing, in general contain elevated concentrations of certain PGE, especially Ir and Rh relative to the chromite-poor rocks of the GNPA member. A high degree of correlation is evident between Rh and Pt (Fig. 5c) and Ir and Ru (Fig. 5f). Broad positive correlations are identifiable between the remaining PGE (Fig. 5a, b, d, and e). Gold shows no relationship with the PGE throughout the chromitites (Fig. 5g, h). Platinum-group elements, Ni and Cu, in general correlate well with each other (Fig. 5a–f and i). The chromitiferous rocks containing significant sulfides (>0.7 wt% S; Table 4) are also relatively Pd-rich with Pt/Pd ratios confined between 0.5 and 0.8. Pt/Pd ratios associated with the sulfide-poor chromitites (<0.3 wt% S; Table 4) are substantially higher ranging between 1.6 and 3.5 (e.g., Fig. 6a). The Pd/Ir (mean of 14) and Rh/Ir (mean of 2) ratios are notably lower in the chromitites than the non chromitiferous rocks.
Geochemical variations with depth
In Fig. 6, borehole RP04.23 provides representative depth profiles of Pt/Pd, Pd/Ir, Rh/Ir, and Ni/Cu ratios within the GNPA member. There is no suggestion that the Pt/Pd, Pd/Ir, and Rh/Ir ratios vary systematically with depth or significantly between the MANO and LMF units (Fig. 6a, b). In contrast, the Ni/Cu ratio decreases slightly with depth (Fig. 6c). This is also reflected in the overall average Ni/Cu ratio of 2 in the MANO unit and 1.2 in the LMF unit. It is important to highlight that the only noticeable variation in the Pt/Pd, Pd/Ir, and Rh/Ir ratios with depth is in association with the chromitite layer (Fig. 6a, b; Table 4).
Chondrite-normalized PGE patterns
Chondrite-normalized PGE patterns for the chromitiferous and non chromitiferous rocks of the GNPA member are shown in Fig. 7. The types of patterns observed are similar to those reported by Maier et al. (2008). The non chromitiferous rocks (Fig. 7a) are characterized by relatively fractionated chondrite-normalized PGE profiles, which peak at Pd. In broad terms, those samples hosting primary and secondary sulfide assemblages exhibit similar shaped profiles; however, within the latter, the profiles are not parallel between Pt, Pd, and Au, which is consistent with the geochemical plots presented in Fig. 5a, e, g, and h. The PGE profiles between the LMF and MANO units are indistinguishable.

Chondrite-normalized PGE profiles for the GNPA member (a) individual profiles for samples containing secondary-bearing sulfides, with the range of primary-bearing sulfide samples also shown (b) chromitites separated into those containing <0.3 wt% S and those with S content >0.7 wt%. Normalization factors from Lodders (2003)
The chromitiferous rocks of the GNPA member are characterized by less fractionated PGE profiles with lower PGE gradients than the non chromitiferous rocks. Two PGE pattern types can be easily identified within the chromitites, which appear to relate directly to sulfur content (Fig. 7b; Table 4). Those chromitites considered S poor (<0.3 wt%) form the characteristic arch-shaped pattern with a peak at Rh or Pt commonly associated with chromite-bearing rocks such as the Merensky Reef and UG2 chromitite (Barnes and Maier 2002a, b; Wilson and Chunnett 2006). The fractionated profiles peak at Rh and contain elevated quantities of IPGE but comparable PPGE concentrations to the non chromitiferous rocks. The second PGE pattern, associated with chromitites containing >0.7 wt% S generally peaks at Pd and is more analogous to those associated with the non chromitiferous rocks.
PGE concentrations in BMS
LA-ICP-MS was utilized to determine the PGE contents of the primary sulfide phases pyrrhotite, pentlandite, and chalcopyrite, and secondary pyrite and millerite, thus providing an insight into the behavior of PGE during low temperature recrystallization and alteration. Results for the laser ablation analysis of sulfides in the non chromitiferous and chromitiferous rocks of the GNPA member are summarized in Table 5. Representative time resolved analysis spectra for those major sulfide phases analyzed, i.e., pentlandite, pyrrhotite, pyrite, and millerite are shown in Fig. 8. All phases carry detectable PGEs in solid solution. As concentrations are very low within chalcopyrite (Table 5), it is not regarded as a significant carrier of PGE within the GNPA member. In general, pyrrhotite, pentlandite, pyrite, and millerite are the major carries of IPGEs, whereas Rh and Pd reside mainly within pentlandite and pyrite, with very low Pt concentrations present in any sulfide phase. Sulfides commonly exhibit zoning of As and Co with elevated concentrations often confined to the boundary of adjacent phases, particularly between pentlandite and pyrite. Abundances of PGEs can be highly variable and erratic both within individual sulfide crystals and sulfide phases.

Selected time resolved analysis spectra for a, b primary pyrrhotite and pentlandite, c composite pentlandite and pyrite, d millerite with PGM, and e, f pyrite and pentlandite from the chromitite
Non chromitiferous rocks
Primary sulfide assemblages
Within the primary assemblages, pyrrhotite and pentlandite were found to carry concentrations of Os (<2 ppm) and Ir (<2 ppm) and higher concentrations of Ru (<18 ppm) in solid solution (Fig. 8a, b). Pyrrhotite in particular shows a high degree of correlation between these elements (Fig. 9a, b). In most samples, pentlandite is slightly more enriched in Os, Ir, and Ru relative to coexisting pyrrhotite (Table 5). This is most apparent for Ru, where concentrations in pentlandite range from 0.05 to <18 ppm, in comparison to <6 ppm in pyrrhotite. Although the Ru content is variable in pentlandite between samples, it is consistent between individual pentlandites within samples. While comparable concentrations of Rh (<5 ppm) are present in pyrrhotite and pentlandite (Fig. 8b), the latter is the principle carrier of Pd with concentrations ranging from 4 to 35 ppm (Fig. 8a, b). Similar to Ru, although the Pd content is rather variable between samples, it is consistent within samples (Fig. 8a). Palladium and Rh show no relationship between one another or with the IPGEs (Fig. 9c, d). No PGEs were present in solid solution or as discrete PGM within chalcopyrite. In contrast to Pd, both Pt and Au are noticeably absent in all the sulfide phases (Table 5).

PGE contents in individual pyrrhotite, pentlandite, pyrite, and millerite grains plotted as a Ir versus Os, b Ir versus Ru, c Ir versus Rh, d Rh versus Pd, e Ir versus Os for chromitites, and f Rh versus Pd for chromitites
Secondary sulfide assemblages
Relicts of primary pyrrhotite within the assemblage pyrite–pentlandite–chalcopyrite ± pyrrhotite ± millerite contain near comparable concentrations of IPGE (all at <1 ppm), Rh, and Pd (both at <2 ppm) in solid solution as within the primary sulfide assemblages. Pentlandite present in secondary textured sulfides is also host to concentrations of Os (<2 ppm), Ir (<1 ppm), and Ru (<9 ppm) in solid solution (Fig. 8c). Ruthenium concentrations show greater variability both between and within samples ranging from below detection limit to 9 ppm (Table 5). Pentlandite remains the principle carrier of Pd and although its content is highly variable between samples (12 to <390 ppm), it is very consistent within samples (Fig. 8c; Table 5). Minor quantities of Rh (<4 ppm) remain present in solid solution within the pentlandite (Fig. 8c). No PGE were present in solid solution within chalcopyrite, with the exception of several analyses, which detected Pd within solid solution at <13 ppm (Table 5).
The most significant relationship found in the secondary sulfides is that pyrite and millerite were found to be important carriers of both Rh and Pd (Fig. 8c, d). Concentrations of both elements in solid solution are highly variable between and within samples (Fig. 8c, d). Palladium ranges from below detection limit to >50 ppm (mean, 7 ppm) in pyrite and millerite (Table 5). Palladium also occurs as discrete PGM (typically Pd–Bi–Te) inclusions within the majority of sulfide phases (e.g., Fig. 8d). The Rh content is slightly elevated within the pyrite (<30 ppm, mean of 4 ppm) relative to coexisting millerite (mean, 2 ppm; Table 5). Palladium and Rh show no relationship between one another or with the IPGEs (Fig. 9c, d). Pyrite and millerite contain low concentrations of Os and Ir (all at <1 ppm), which, like in the primary phases, correlate well with each other (Fig. 9a). Ruthenium concentrations are slightly higher and more variable within pyrite (<10 ppm) relative to millerite, but a strong correlation with Ir is still preserved. Platinum, in contrast to Pd, is noticeably absent in the majority of phases only being detected in solid solution and as occasional PGM (Pt–Bi–Te) within several pyrite and pentlandite analyses (Fig. 8c; Table 5). Pyrite also contains gold in solid solution at concentrations of <2 ppm. These concentrations are notably higher than observed in the other sulfide phases within the GNPA member (Table 5).
Chromitiferous rocks
The chromitiferous rocks of the GNPA member contain significantly elevated concentrations of IPGE in comparison to the non chromitiferous rocks (Tables 4 and 5). Pyrite is the principal carrier of the IPGEs, where concentrations of Os (<14 ppm; mean, 1.5 ppm), Ir (<10 ppm; mean, 1 ppm), and Ru (<124 ppm; mean, 13 ppm) are highly irregular between samples and within individual grains (Fig. 8e). The IPGEs are also found in solid solution within millerite, pentlandite, and chalcopyrite but at lower and more consistent concentrations (Fig. 8e; Table 5). There is a high degree of correlation within all phases between the IPGEs (Fig. 9e).
Rhodium is hosted in solid solution principally by pyrite with lower concentrations identified within pentlandite and millerite (<5 ppm; Table 5; Fig. 8e, f). Interestingly, As and Bi exhibit parallel profiles to Rh (Fig. 8f), which may result from the zonation of these elements. Within pyrite, the Rh content is highly irregular reaching up to 54 ppm with a mean of only 5 ppm (Fig. 8f). Palladium was detected in all sulfide phases with the exception of chalcopyrite (Table 5). Pentlandite is the principal carrier of Pd (Fig. 8e, f), with concentrations being highly erratic (0.9–192 ppm; mean, 88 ppm) even within a single grain. Pyrite is also an important host, although concentrations are lower at <16 ppm and highly varied (mean, 2 ppm; Fig. 8e, f). Millerite contains only minor quantities of Pd at <0.6 ppm (Fig. 8d). Correlations between Pd and Rh (Fig. 9f) and between Pd and IPGEs are poor. Similar to the non chromitiferous rocks, Pt is generally absent from the majority of phases or present at very low concentrations in solid solution and as occasional PGM. Concentrations of Pt (up to 9 ppm) were found in a few analyses of pyrite and pentlandite, but the majority of analyses found no Pt above the limit of detection (Table 5).
Mass balance
For a semi-quantitative indication of the proportion of PGE present within BMS and how these proportions change as the BMS assemblage changes, we performed a mass balance, following similar methods to Huminicki et al. (2005) and Holwell and McDonald (2007).
For the primary pyrrhotite–pentlandite–chalcopyrite sulfide assemblage, we applied a similar approach to Holwell and McDonald (2007). As chalcopyrite contains virtually no PGE in solid solution, we recalculated the whole-rock PGE contents to 100 % pyrrhotite and pentlandite as these are the principle phases that contain PGE in solid solution. To determine the weight fraction of the sulfide phases present, we utilized the method of Huminicki et al. (2005), using whole-rock Cu, Ni, and S data. All whole-rock Cu was assigned to chalcopyrite, and Ni was assigned to pentlandite, following a correction to account for trace amounts of Ni in pyrrhotite and silicates. The proportion of pyrrhotite was then obtained assuming that the remaining S, after subtracting the S required by pentlandite and chalcopyrite, corresponds to pyrrhotite.
Where the secondary pyrite–chalcopyrite–pentlandite ± millerite ± pyrrhotite assemblage is developed, the presence of pyrite and millerite was also taken into account. As no samples used in the calculation contained both millerite and pentlandite, we assigned all whole-rock Ni to the mineral present. The weight fraction of pyrite was then obtained assuming all remaining S corresponds to pyrite. This assumes no pyrrhotite, but is valid in this case as no pyrrhotite was observed in the samples used in the mass balance.
In Fig. 10, we compare the average PGE contents of pyrrhotite, pentlandite, pyrite, and millerite determined by LA-ICP-MS, normalized to chondrite, and the mean whole-rock concentrations of PGE and Au for the same samples, recalculated in 100 % sulfide. This method is adapted from Ballhaus and Sylvester (2000), with the rationale that an element whose concentration within a sulfide is as high or higher than the recalculated whole-rock contents indicates its presence in solid solution, whereas if it falls below, some of that element must be present as other discrete phases.

Chondrite-normalized diagrams of average PGE in a pentlandite and bulk sulfide recalculated to 100 % sulfide (po + pn and pn only) for primary sulfide-bearing rocks, b pyrrhotite and bulk sulfide recalculated to 100 % sulfide (po + pn and po only) for primary sulfide-bearing rocks, c pentlandite and bulk sulfide in 100 % sulfide (py + pn + mill and pn only) for secondary sulfide-bearing rocks, and d pyrite and bulk sulfide in 100 % sulfide (py + pn + mill and py only) also for secondary sulfide-bearing rocks
Primary sulfide assemblages
The IPGE are accommodated comfortably in solid solution within pyrrhotite and pentlandite (Fig. 10a, b) as averaged IPGE concentration of both sulfide phases, plot higher than the recalculated bulk rock contents. The elevated concentration of IPGE in both phases, relative to whole rock, indicates that these elements are present primarily within one phase (Fig. 10a, b). When whole rock is recalculated to 100 % pentlandite (Fig. 10a) and 100 % pyrrhotite (Fig. 10b), it is clear Ir and Ru are primarily present within pyrrhotite as whole-rock Ir and Ru is almost identical to that in pyrrhotite (Fig. 10b). Rhodium and Pd both fall slightly below whole-rock values indicating they must also be present as discrete PGM. The large negative anomalies in both Pt and Au show that these are the only metals to reside primarily as discrete phases with only a small fraction being held in solid solution. These results are in agreement with our PGM study, which identified >70 % of all PGM (by area) to be Pt phases, around 20 % Pd, 2 % Au, and only 1 % to be Rh phases.
Secondary sulfide assemblages
Large discrepancies exist between the observed data (both LA-ICP-MS and PGM studies) and that calculated by the mass balance as the averaged laser data is significantly lower, by an order of magnitude, to that recalculated from whole rock. The mass balance (Fig. 10c, d) suggests that Os, Ir, and Ru occur primarily as discrete PGM phases where secondary sulfides exist. This seems highly implausible mainly because no Os-, Ir-, or Ru-bearing PGM have been identified and the bulk PGE data (Fig. 5d, f) implies that these elements were not remobilized during alteration. Thus, the IPGE are expected to remain hosted by BMS, as seen in the primary sulfide assemblage mass balance (Fig. 10a, b). According to the mass balance, Pd is fully accommodated within pentlandite; however, this is not consistent with our PGM study which identified 41 % of all PGM by area to be Pd phases. There are a number of reasons why this mass balance may not be an accurate representation of the mineralization present. Firstly, the secondary sulfide assemblages are substantially more complex than portrayed in the calculation and vary significantly between samples. Furthermore, within fluid affected ore-bodies S-loss is common. Not correcting for this loss will effectively result in the PGE being greatly concentrated in the calculated sulfide fractions, thus resulting in a large discrepancy between the observed and calculated PGE contents as evident in Fig. 10c and d.
Overall, it is evident that our mass balance works well for samples hosting primary sulfides and is thus in these instances an accurate representation of the mineralization present within the GNPA member prior to alteration. In contrast, due to the many variables and unknowns, our mass balance cannot be used with any degree of certainty for those samples hosting secondary sulfides.
Discussion
Our data show that differences in the geochemical and mineralogical characteristics of PGE and BMS mineralization within the GNPA member correlate well with sulfide assemblage type and are thus controlled by magmatic and hydrothermal processes. Significant features identified within this study include (1) the strong correlation between PGE, S, and base metals in primary sulfide assemblages; (2) variation of platinum-group mineralogy between sulfide assemblages; (3) the dominance of sulfide PGE patterns in sulfide-rich chromitites; (4) the presence of IPGE, Pd and Rh within pyrite and millerite; and (5) the lack of correlation between Pd and Au with Pt and Ir in fluid-affected zones. In the following discussion, we investigate the genetic implications of these features through applying our data to the current suggested models for the potential correlative Platreef and Merensky Reef. We start, therefore, to constrain the mechanisms involved in the development of GNPA mineralization and explore the behavior of PGE during both initial sulfide fractionation and low temperature recrystallization.
Primary magmatic signature
The development of a primary sulfide liquid throughout the GNPA member is supported by the strong correlation evident between the chalcophile elements and S within the primary sulfide assemblage, which indicates the initial concentration of these elements, was governed by a single sulfide melt. This is further supported by the similarity of the associated, Bi–Te–As dominated PGM assemblage and the consistency of the Pt/Pd, Pd/Ir, and Rh/Ir ratios throughout the entire GNPA stratigraphy (Fig. 6a, b; Table 4) as all imply crystallization from a compositionally similar PGE-rich sulfide liquid.
This study has also revealed that where primary sulfides exist: (1) all IPGE and Rh occur in solid solution within pyrrhotite and pentlandite (Fig. 10a); (2) pentlandite is a significant host of Pd with the rest occurring as PGM; (3) Pt resides primarily as discrete PGM; and (4) PGM are located in association with sulfides (Table 3). All these observation are consistent with the fractionation and crystallization of a magmatic sulfide liquid (Cabri and Laflamme 1976; Fleet et al. 1993; Li et al. 1996; Ballhaus et al. 2001; Mungall et al. 2005; Barnes et al. 2006; Holwell and McDonald 2010; McDonald and Holwell 2011). The presence of IPGE and Rh within pyrrhotite and pentlandite is consistent with the exsolution of these phases from early crystallizing monosulfide solid solution (mss), with which these elements are highly compatible with (Barnes et al. 2006). Platinum, Pd, and Au are considered incompatible within both mss and intermediate solid solution (iss), which crystallizes from the residual fractionated sulfide liquid (Fleet et al. 1993; Li et al. 1996; Peregoedova 1998). These elements are therefore preferentially concentrated into a late-stage immiscible semi-metal-rich melt (Fleet et al. 1993; Helmy et al. 2007, 2010; Tomkins 2010). Where semi-metals are in abundance (particularly Sb and As), through contamination at high temperatures (e.g., Platreef at Turfspruit), virtually all the Pt and Pd can be accommodated within the semi-metal-rich melt and thus reside as PGM (e.g., Hutchinson and McDonald 2008). In contrast, where semi-metals have been sourced directly from the magma and are thus limited (e.g., Platreef at Overysel), the late-stage melt preferentially scavenges Pt over Pd (Fleet et al. 1993; Helmy et al. 2007). This is observed within the GNPA member and thus provides evidence that, prior to sulfide immiscibility, the magma had not been significantly contaminated specifically with semi-metals. In this situation, the presence of a high Pd/semi-metal ratio results in excess Pd, which cannot be accommodated for within the semimetal melt, to partition into mss (Helmy et al. 2007). The presence of Pd in pentlandite is a feature also observed in the Platreef and many other Ni–Cu–PGE deposits (e.g., Cabri et al. 1984; Czamanske et al. 1992; Ballhaus and Ryan 1995; Godel et al. 2007; Holwell and McDonald 2007; Djon and Barnes 2012), where it is interpreted to result from Pd preferentially diffusing into pentlandite over pyrrhotite during recrystallization of mss. Within the GNPA member the primary sulfide associated Pt–As and Pd–Bi–Te dominated PGM assemblage (Tables 1 and 2) crystallized around the margins of sulfides, as the semi-metal melt was expelled to grain boundaries during crystallization of iss in the manner described by Holwell and McDonald (2010). Later replacement, around the margins of the sulfide blebs by secondary actinolite, tremolite, and chlorite (Smith et al. 2011) isolates the PGMs as satellite grains within secondary silicates, which is a feature common throughout the GNPA member.
In addition to sulfide liquid, chromite precipitation is also known to effectively concentrate PGE, especially IPGE and Pt (see von Gruenewaldt et al. 1989: Barnes and Maier 2002a, b; Prichard et al. 2004; Godel et al. 2007). Where this mechanism of PGE enrichment prevails, chromitites are characterized by: Pt/Pd > 1, arched chondrite-normalized PGE profiles, and an increase in PGE sulfides and sulfarsenides (Kinloch 1982; Kinloch and Peyerl 1990; Barnes and Maier 2002a, b; Wilson and Chunnett 2006). Since chromitites within the GNPA member are characterized by either a chromite or sulfide signature, we believe both mechanisms of PGE enrichment were in operation within the parental magma (Table 4 and Fig. 7). Based on the key observations that the chromite signature is confined to those chromitites considered S poor (<0.3 wt% S and Fig. 7b) and elevated grades occur in association with the S-rich chromitites (>0.7 wt% S; Fig. 7b and Tables 1, 2, and 4), we infer that where present sulfides were the main control over bulk PGE grades and relative element ratios within the GNPA member. Within the chromitites, we believe PGE enrichment to have occurred in two stages: (1) some IPGE and Pt were concentrated during chromite precipitation, with the presence of PGE alloys in association with the chromitite layer at War Springs (Fig. 1; Sutherland 2013) being indicative of such conditions and (2) with the remaining PGE collected by an immiscible sulfide liquid. It may be possible that if the chromite (and any associated Pt-rich PGM) had become mixed with any subsequent sulfide liquid the initial Pt-rich character may have been overprinted or lost if the ratio of sulfide to chromite was sufficiently high. Where sulfides did not significantly interact with the chromitite layer, chromite was the principal mechanism by which PGE were concentrated thus high Pt/Pd ratios (>1), reflecting the preferential fractionation of Pt over Pd by chromite (Barnes and Maier 2002b), and associated platinum-group mineralogy (Tables 1, 2, and 4) are preserved. Our observations imply that within the GNPA member the magma (s) from which chromite crystallized had not been depleted of its PGE (in particular Pt, Rh, and IPGE) contents prior to the formation of chromite.
Hydrothermal interaction
The most striking difference between the PGM assemblages in the primary and secondary sulfides is the greater abundance of Sb-bearing PGM (e.g.. stibiopalladinite and sudburyite) in association with the hydrothermally altered sulfides (Tables 1 and 2). The occurrence of significant quantities of Pd antimonides and Pd arsenides is considered indicative of either hydrothermal interaction (e.g., Cabri et al. 2005; McDonald et al. 2005a, b; Holwell et al. 2006, 2014), or contamination (e.g. Hutchinson and Kinnaird 2005; Hutchinson and McDonald 2008). Within the GNPA member, we believe that fluids interacted with the primary sulfide and associated PGM assemblage resulting in the direct alteration of the PGE and sulfide mineralogy. Although the mineralogy of some PGM has changed, they continue to reside in close association with the sulfides (Table 3), thus indicating recrystallization occurred in situ with minimal remobilization of PGE. This could directly result from the high quantities of Sb, As, Bi, and Te believed to have been present within the volatile phase, as these act to restrict the mobility of PGE rather than facilitate transportation of them (Mountain and Wood 1988).
Within the Platreef, volatile-rich fluids are thought to originate from metasedimentary crustal xenoliths and metamorphism of footwall dolomite and shale (Sharman-Harris et al. 2005; Holwell and McDonald 2006; Holwell et al. 2006; Pronost et al. 2008). Since the footwall to the GNPA member consists of quartzite and Lower Zone cumulates, we suggest that Sb-bearing fluids were derived from calc-silicate xenoliths, up to several meters in thickness, which have been identified along the footwall contact and within the GNPA member (Maier et al. 2008). Although dolomites do not form the immediate footwall to the GNPA member, the presence of calc-silicates may suggest that they were assimilated by the GNPA magma in a downdip direction (Maier et al. 2008).
The behaviour of PGE during low temperature alteration
A major finding of this study is the presence of significant quantities of PGE held in solid solution within pyrite, a feature also documented within other Ni–Cu–PGE sulfide deposits (e.g., Oberthür et al. 1997; Barkov et al. 1997; Gervilla and Kojonen 2002; Cabri et al. 2002, 2008, 2010; Djon and Barnes 2012; Dare et al. 2011; Piña et al. 2012, 2013). Within the GNPA member, the pyrite is (1) host to comparable concentrations of IPGEs as pyrrhotite and pentlandite (Fig. 8b; Table 5), (2) significantly enriched in Rh (≥54 ppm; Fig. 8b and Table 5), and (3) considerably lower in Pd contents than pentlandite (Fig. 8b and Table 5). These observations imply that the pyrite most likely directly inherited its PGE contents from the pyrrhotite and pentlandite it replaced during low temperature alteration, in a similar manner to that proposed by Dare et al. (2011) and Djon and Barnes (2012) for the McCreedy East and Lac des Iles deposits, respectively. The comparable concentrations of IPGE in pyrrhotite, pentlandite, and pyrite further highlight the immobile manner of these elements during sulfide replacement within the GNPA member. In contrast, the lower concentrations of Pd (<100 ppm) in pyrite than typical of pentlandite (>100 ppm) being directly replaced is consistent with the geochemical data, indicating Pd has experienced partial remobilization during low temperature alteration (Fig. 8b). Our data also highlight the ability of pyrite to host appreciable concentrations of Rh (≥54 ppm; Table 5). The mechanisms by which Rh becomes concentrated within secondary pyrite are at present not well understood. Our study revealed that millerite also hosts PGE in solid solution. Although concentrations of Pd, Rh, and IPGE are typically lower than within pyrite, we suggest that millerite also directly inherited its PGE contents from the phases it replaced.
Evaluation of ore forming processes
In starting to constrain the mechanisms involved in the formation of the mineralization within the GNPA member, we explore two genetic models that are attributed to the generation of PGE mineralization within the Merensky Reef and the Platreef. These are, respectively:
-
1.
Sulfide saturation during emplacement—extraction of PGE from new magma influx
-
2.
Sulfide saturation in a staging chamber, with emplacement of pre-formed PGE rich sulfides
In the following discussion, we apply our data for the GNPA member to these two models, thus highlighting any potential common ore-forming processes and providing possible constraints on the timing of S saturation relative to emplacement.
Sulfide saturation during emplacement
The Merensky Reef represents a stratiform type deposit, formed principally by the settling of a dense, immiscible sulfide liquid through a column of S-saturated magma (see Barnes and Maier 2002a, b and references therein). Mixing of residual and primitive (Main Zone) magmas is thought to have initially induced S saturation, consequently depleting the magma of its metals (Maier and Barnes 1999; Li and Ripley 2005). The Main Zone within the eastern and western limbs of the Bushveld Complex is therefore depleted of PGE and is considered unprospective. In the case of the GNPA member, we strongly believe that this model is not applicable for the following reasons. In recent years, it has become apparent that the Main Zone within the northern limb is also host to PGE mineralization (Maier and Barnes 2010; McDonald and Harmer 2011; Lombard 2012; Kinnaird et al. 2012; Holwell et al. 2013). On the farm Moorddrift (Figs. 1 and 2), Holwell et al. (2013) describes the sulfide associated mineralization as stratiform type reefs, which are magmatic in origin and thus unrelated to the underlying GNPA member or Platreef. These observations, in conjunction with the identification of a magmatic break between the intrusion of the Platreef and Main Zone (Holwell et al. 2005; Holwell and Jordaan 2006), suggest that the Main Zone was emplaced as a fertile magma with a separate PGE budget from the underlying deposits. In addition, within the GNPA member PGE and BMS mineralization is hosted only within the LMF and MANO units, with the separating LGN unit (Fig. 2) being completely barren. De Klerk (2005) proposed that the LGN unit represents a sill of Main Zone; if this is accepted, then it would suggest that an immiscible sulfide liquid was developed within both the LMF and MANO units prior to intrusion of the Main Zone. All of the above evidence strongly implies that (1) the GNPA member did not source its PGE in situ from the overlying Main Zone and (2) that S saturation occurred prior to emplacement of Main Zone.
Another feasible mechanism by which S saturation can be reached during emplacement is through in situ contamination. The addition of crustal S through assimilation of S-bearing country rocks is considered by many as being an essential process in the generation of large magmatic ore deposits (Lesher and Groves 1986). Where contamination occurs in situ, sulfide mineralization is typically developed along the basal parts of the intrusion (e.g., Duluth Complex; Mainwaring and Naldrett 1977; Ripley 1981; Ripley et al. 1986; and the Basal Series of the Stillwater Complex; Lee 1996; McCallum 1996). Within the GNPA member, however, mineralization is not restricted along its basal margin, being observed throughout the entire 400–800 m thick succession. In addition, this genetic model becomes more unfeasible for the GNPA member when the local country rocks are considered (Figs. 1, 2, and 3). West of the Grasvally Fault, the footwall consists of 800–1,600 m succession of Lower Zone cumulates (Figs. 1 and 2); if contamination was local and in situ, then sulfide mineralization would not be expected to be developed throughout the GNPA member within this area. Furthermore, east of this fault the GNPA member is underlain by quartzites from the Magaliesberg Quartzite Formation, which are an unlikely source of crustal S as they do not contain significant quantities of S (Smith et al. 2013).
In addition, the restriction of Sb-bearing PGM to those sulfides, which have experienced hydrothermal alteration, is also inconsistent with this model. Hutchinson and Kinnaird (2005) and Hutchinson and McDonald (2008) highlighted that along with S, semi-metals (particularly Sb) are also introduced into the magma and sulfide liquid during assimilation of local country rocks (e.g., Platreef at Turfspruit). Therefore, within the GNPA member, one would expect the primary sulfide associated PGM assemblage to also be abundant in Sb-bearing PGM. Finally, preliminary S isotope results provide no indication that the GNPA member experienced local contamination during or post emplacement but does reveal that the magma was extensively contaminated with crustal S (Smith et al. 2012, 2013). In considering all the evidence presented, it appears highly implausible that the parental magma (s) of the GNPA member became S saturated during emplacement, thus a model that enables the magma to be both S saturated and PGE-rich at the time of emplacement is more favorable.
Staging chamber model
It is generally accepted that the Platreef was emplaced as a number of sills that already contained a PGE-enriched sulfide liquid (e.g., Lee 1996; Kinnaird 2005; Holwell et al. 2007; McDonald and Holwell 2007). In the current model, early-stage contamination induced sulfide immiscibility at depth prior to emplacement (Ihlenfeld and Keays 2011). The early-formed sulfide liquid subsequently became progressively enriched in PGE, Ni, and Cu through reacting with multiple batches of silicate magma at low R factors. Subsequent to further upgrading by partial dissolution (in the manner described by Kerr and Leitch 2005), the sulfides were remobilized and emplaced into the Platreef (McDonald and Holwell 2011; McDonald et al. 2012).
The stratigraphic setting of the GNPA member is analogous to that of the adjacent Platreef. In the discussion above, we suggested that the GNPA member in essence requires a genetic model comparable to that proposed for the Platreef. The Lower Zone cumulates that directly underling the GNPA member are PGE, Ni, and Cu depleted (McDonald and Holwell 2007), a feature considered to be consistent with the enrichment of sulfides through processing of pre-GNPA magma(s) within a deeper magmatic system (see also McDonald et al. 2009). This therefore implies that the GNPA member may have sourced its PGE content from the magma, which was intruded to form the underlying Lower Zone. The involvement of a deeper chamber is further supported by the S isotope evidence, which suggests the GNPA member was extensively contaminated with crustal S (Smith et al. 2012, 2013), a feature not indicative of the in situ assimilation of S-bearing country rocks. Due to the lack of local S-bearing country rocks, this must have occurred in a deeper magmatic system.
If the Platreef genetic model is applied to the GNPA member, then the development of elevated PGE tenors only within the Platreef (Holwell and McDonald 2007) must be plausibly accounted for. The lower PGE tenors of sulfides obtained through our LA-ICP-MS data associated with the GNPA member are interpreted to be a primary feature, as the current study highlights that hydrothermal fluids have not significantly redistributed PGE from BMS. Consequently, the presence of lower tenors in the GNPA member may be ascribed to: its generation from magma poorer in PGE, interaction of sulfides with a smaller volume of magma compared to the Platreef, or dilution of the PGE content within sulfide prior to emplacement. Additionally, within the Platreef staging chamber partial dissolution of sulfides contributed to the development of high PGE tenors (McDonald et al. 2012) that are comparable to those in the Merensky Reef (Godel et al. 2007). Therefore, it is also possible that this process of upgrading was not in operation within the system, which supplied the GNPA member, resulting in sulfides appearing poorer in PGE. Although these suggestions still enable the GNPA member to correspond with the Platreef, this discussion raises the possibility that: (1) the GNPA and Platreef were derived from magma differing slightly in composition, particularly in terms of PGE content and/or (2) that the parental magmas and PGE-rich sulfides of the GNPA member and the Platreef were supplied from a complex network of chambers and conduits, where the degree of sulfide dissolution and PGE enrichment was variable.
In summary, from the data currently available, we propose that the GNPA member was emplaced in a similar manner to the Platreef involving the development of a sulfide liquid, enriched in PGE by equilibrating with a large volume of magma at depth in a conduit system. At present, the importance of early-stage crustal contamination in driving S saturation can only be speculated and will only be revealed through application of other techniques such as S isotopes and S/Se ratios. We envisage that the GNPA member most probably formed within the same conduit network as the Platreef, notable differences however in PGE tenor suggests that different ore forming processes operated north and south of the Ysterberg–Planknek Fault.
Conclusions
This study has revealed that the distribution of platinum-group and chalcophile elements within the GNPA member results from the complex behavior of these elements during both magmatic and hydrothermal processes. The distribution of PGE within the primary sulfide assemblage and associated Pt–As and Pd–Bi–Te dominant PGM assemblage is consistent with the fractionation of a single sulfide liquid. Post emplacement fluid interaction has resulted in the decoupling of Pd, Au, and Cu from sulfides on a centimeter to decimeter scale, and the development of a more Sb-bearing PGM assemblage, characteristic of hydrothermal fluids. Recrystallization of PGM and sulfides occurred in situ, resulting in pyrite and millerite inheriting PGE directly from the pyrrhotite and pentlandite replaced. We reveal therefore that pyrite and millerite can be important carriers of IPGE, Rh, and Pd.
In starting to constrain the ore genesis of sulfide and associated PGE mineralization within the GNPA member, we reject any model where sulfide immiscibility was induced during or post emplacement and thus through either in situ contamination or depletion of an overlying magma column by a settling sulfide liquid. We therefore favor a model similar to that proposed for the Platreef, where PGE-rich sulfides were formed at depth in a conduit system prior to emplacement. It is not yet clear how the GNPA member relates to the Platreef, although it is likely that they formed within the same conduit network. Notable differences in PGE tenor suggests that the processes involved in ore formation and PGE-enrichment may have differed within the parental magmas of the GNPA member and the Platreef.
References
Armitage PEB, McDonald I, Edwards SJ, Manby CM (2002) Platinum-group element mineralization in the Platreef and calc-silicate footwall at Sandsloot, Potgietersrus district, South Africa. Appl Earth Sci (Trans Inst Min Metall B) 111:B36–B45
Ballhaus C, Ryan CG (1995) Platinum-group elements in the Merensky Reef. I. PGE in solid solution in base metal sulfides and the down-temperature equilibration history of Merensky ores. Contrib Mineral Petrol 122:241–251
Ballhaus C, Sylvester P (2000) Noble metal enrichment processes in the Merensky Reef, Bushveld Complex. J Petrol 41:545–561
Ballhaus C, Tredoux M, Spath A (2001) Phase relations in the Fe–Ni–Cu–PGE–S system at magmatic temperature and application to massive sulphide ores of the Sudbury Igneous Complex. J Petrol 42:1911–1926
Barkov AY, Halkoaho TAA, Laajoki KVO, Alapieti TT, Peura RA (1997) Ruthenian pyrite and nickeloan malanite from the Imandra layered complex, northwestern Russia. Can Mineral 35:887–897
Barnes S-J, Maier WD (2002a) Platinum-group element distributions in the Rustenburg Layered Suite of the Bushveld Complex, South Africa. Can Inst Min Metall Pet 54:553–580
Barnes S-J, Maier WD (2002b) Platinum-group elements and microstructures of normal Merensky Reef from Impala Platinum Mines, Bushveld Complex. J Petrol 43:103–128
Barnes S-J, Cox RA, Zeintek ML (2006) Platinum-group element, gold, siliver and base metal distribution in compositionally zoned sulfide droplets from the Medvezky Creek Mine, Noril’sk, Russia. Contrib Mineral Petrol 152:187–200
Buchanan PC, Reimold WU, Koeberl C, Kruger FJ (2002) Geochemistry of intermediate to siliceous volcanic rocks of the Rooiberg Group Bushveld Magmatic Province South Africa. Contrib Mineral Petrol 144:131–143
Cabri LJ, Laflamme JHG (1976) The mineralogy of the platinum-group elements from some copper–nickel deposits of the Sudbury area, Ontario. Econ Geol 71:1159–1195
Cabri LJ, Bank H, El Goresy A, Laflamme JHG, Nobiling R, Sizgoric MB, Traxel K (1984) Quantitative trace-element analyses of sulfides from Sudbury and Stillwater by proton microprobe. Can Mineral 22:521–542
Cabri LJ, Wilson JMD, Distler VV, Kingston D, Nejedly Z, Sluzheniken SF (2002) Mineralogical distribution of trace platinum-group elements in the disseminated sulphide ores of Norilsk 1 layered intrusion. Appl Earth Sci (Trans Inst Min Metall B) 111:B15–B22
Cabri LJ, McDonald AM, Stanley CJ, Rudashevsky NS, Poirier G, Durham BR, Mungall JE, Rudashevsky VN (2005) Naldrettite, Pd2Sb, a new palladium antimonide from the Mesamax Northwest deposit, Ungava region, Québec, Canada. Mineral Mag 69:89–97
Cabri LJ, Rudashevsky NS, Rudashevsky VN (2008) Current approaches for the process mineralogy of platinum-group element ores and tailings. In: Ninth international congress for Applied Mineralogy ICAM 2008. The Australasian Institute of Mining and Metallurgy, publication series no 8/2008, pp 9–17
Cabri LJ, Choi Y, Nelson M, Tubrett M, Sylvester PJ (2010) Advances in precious metal trace element analyses for deportment using LAM-ICP-MS. In: Proceedings of the 42nd Annual Canadian Mineral Processors Conference, pp 181–196
Cawthorn RG, Davies G, Clubley-Armstrong A, McCarthy TS (1981) Sills associated with the Bushveld Complex. Lithos 14:1–15
Czamanske GK, Kunilov VE, Zientek ML, Cabri LJ, Likhachev AP, Calk LC, Oscaron RL (1992) A proton-microprobe study of magmatic sulfide ores from the Noril’sk-Talnakh district, Siberia. Can Mineral 30:249–287
Dare SAS, Barnes S-J, Prichard HM, Fisher PC (2011) Chalcophile and platinum-group element (PGE) concentrations in the sulfide minerals from the McCreedy East deposit, Sudbury, Canada, and the origin of PGE in pyrite. Mineral Deposita 46:381–407
de Klerk L (2005) Bushvled Stratigraphy on Rooipoort, Potgietersrus Limb. Platreef Workshop, 2nd, Mokopane, South Africa, 28th–30th October 2005, Abstracts
Djon MLN, Barnes S-J (2012) Changes in sulphides and platinum-group minerals with the degree of alteration in the Roby, Twilight, and High Grade Zones of the Lac des Iles Complex, Ontario, Canada. Mineral Deposita 47:875–896
Dunnett T, Grobler DF, Simmonotti NMEM, Mapeka JM (2012) Lithological variations within Upper Critical Zone stratigraphy, Turfspruit 241KR, northern limb, Bushveld Complex. Platreef Workshop, 5th, Mokopane, South Africa, 9th – 11th November 2012, Abstracts
Eales HV, Cawthorn RG (1996) The Bushveld Complex. In: Cawthorn RG (ed) Layered intrusions. Elsevier Science, Amsterdam, pp 181–230
Fleet ME, Chryssoulis SL, Stone WE, Weisener CG (1993) Partitioning of platinum-group elements and Au in the Fe–Ni–Cu–S system: experiments on the fractional crystallization of sulfide melt. Contrib Mineral Petrol 115:36–44
Gervilla F, Kojonen K (2002) The platinum-group minerals in the upper section of the Keivitsansarvi Ni–Cu–PGE deposit, northern Finland. Can Mineral 40:377–394
Godel B, Barnes S-J, Maier WM (2007) Platinum-group elements in sulfide minerals, platinum-group minerals, and whole-rocks of the Merensky Reef (Bushveld Complex, South Africa) Implications for the formation of the reef. J Petrol 48:1569–1604
Grobler DF, Nielsen SA, Broughton DW (2012) Upper Critical Zone (Merenksy Reef) correlates within the Platreef on Turfspruit 241KR, northern limb, Bushveld Complex. Platreef Workshop, 5th, Mokopane, South Africa, 9th – 11th November 2012, Abstracts
Harris C, Chaumba JB (2001) Crustal contamination and fluid-rock interaction during the formation of the Platreef, northern lobe of the Bushveld Complex, South Africa. J Petrol 42:1321–1347
Helmy HM, Ballhaus C, Berndt J, Bockrath C, Wohlgemuth-Ueberwasser C (2007) Formation of Pt, Pd and Ni tellurides; experiments in sulfide-telluride systems. Contrib Mineral Petrol 153:577–591
Helmy HM, Ballhaus C, Wohlgemuth-Ueberwasser C, Fonseca ROC, Laurenz V (2010) Partitioning of Se, As, Sb, Te and Bi between monosulfide solid solution and sulfide melt—application to magmatic sulfide deposits. Geochim Cosmochim Acta 74:6174–6179
Holwell DA, Jordaan A (2006) Three-dimensional mapping of the Platreef at the Zwartfontein South mine: implications for the timing of magmatic events in the northern limb of the Bushveld Complex, South Africa. Appl Earth Sci (Trans Inst Min Metall B) 115:B41–B48
Holwell DA, McDonald I (2006) Petrology, geochemistry and the mechanisms determining the distribution of platinum-group element and base metal sulfide mineralization in the Platreef at Overysel, northern Bushveld Complex, South Africa. Mineral Deposita 41:575–598
Holwell DA, McDonald I (2007) Distributions of platinum-group elements in the Platreef at Overysel, northern Busveld Complex: a combined PGM and LA-ICP-MS study. Contrib Mineral Petrol 154:171–190
Holwell DA, McDonald I (2010) A review of the behaviours of platinum-group elements within natural magmatic sulfide ore systems. Platin Met Rev 54:26–36
Holwell DA, Armitage PEB, McDonald I (2005) Observations on the relationship between the Platreef and its hangingwall. Appl Earth Sci (Trans Inst Min Metall B) 114:B225–B241
Holwell DA, McDonald I, Armitage PEB (2006) Platinum-group mineral assemblages in the Platreef at the Sandsloot Mine, northern Bushveld Complex, South Africa. Mineral Mag 70:83–101
Holwell DA, Boyce AJ, McDonald I (2007) Sulfur isotope variations within the Platreef Ni–Cu–PGE deposit: genetic implications for the origin of sulfide mineralization. Econ Geol 102:1091–1110
Holwell DA, McDonald I, Butler IB (2011) Precious metal enrichment in the Platreef, Bushveld Complex, South Africa: evidence from homogenized magmatic sulfide melt inclusions. Mineral Deposita 161:1011–1026
Holwell DA, Jones A, Smith JW, Boyce AJ (2013) New mineralogical and isotopic constraints on Main Zone-hosted PGE mineralisation at Moorddrift, northern Bushveld Complex. Mineral Deposita 48:675–686
Holwell DA, Keays RR, Firth EA, Findlay J (2014) Geochemistry and mineralogy of platinum-group element mineralization in the River Valley intrusion, Ontario, Canada: a model for early stage S saturation and multi-stage emplacement and the implications for ‘contact-type’ Ni-Cu-PGE mineralization. Econ Geol 109:689–712
Huber H, Koeberl C, McDonald I, Reimold WU (2001) Geochemistry and petrology of Witwatersrand and Dwyka diamictites from South Africa: search for an extra-terrestrial component. Geochim Cosmochim Acta 65:2007–2016
Hulbert LJ (1983) A petrographical investigation of the Rustenburg Layered Suite and associated mineralization south of Potgietersrus. D.Sc. dissertation, Pretoria, South Africa, The University of Pretoria
Huminicki MAE, Sylvester PJ, Cabri LJ, Lesher CM, Tubrett M (2005) Quantitative mass balance of platinum group elements in the Kelly Lake Ni–Cu–PGE deposit, Copper Cliff offset, Sudbury. Econ Geol 100:1631–1646
Hutchinson D, Kinnaird JA (2005) Complex multistage genesis for the Ni-Cu-PGE mineralization in the southern region of the Platreef, Bushveld Complex, South Africa. Appl Earth Sci (Trans Inst Min Metall B) 114:B208–B223
Hutchinson D, McDonald I (2008) Laser ablation ICP-MS study of platinum-group elements in sulfides from the Platreef at Turfspruit, northern limb of the Bushveld Complex, South Africa. Mineral Deposita 43:695–711
Ihlenfeld C, Keays R (2011) Crustal contamination and PGE mineralization in the Platreef, Bushveld Complex, South Africa: evidence for multiple contamination events and transport of magmatic sulfides. Mineral Deposita 46:813–832
Keays RR, Nickel EH, Groves DI, McGoldrick PJ (1982) Iridium and palladium as discriminants of volcanic exhalative, hydrothermal and magmatic nickel sulfide mineralization. Econ Geol 77:1535–1547
Kerr A, Leitch AM (2005) Self destructive sulfide segregation systems and the formation of high-grade magmatic ore deposits. Econ Geol 100:311–332
Kinloch ED (1982) Regional trends in the platinum-group mineralogy of the critical zone of the Bushveld Complex, South Africa. Econ Geol 77:1328–1347
Kinloch ED, Peyerl W (1990) Platinum-group minerals in various rock types of the Merensky Reef: genetic implications. Econ Geol 85:537–555
Kinnaird JA (2005) Geochemical evidence for multiphase emplacement in the southern Platreef. Appl Earth Sci (Trans Inst Min Metall B) 114:B225–B241
Kinnaird JA, McDonald I (2005) An introduction to mineralisation in the northern limb of the Bushveld Complex. Appl Earth Sci (Trans Inst Min Metall B) 114:B194–B198
Kinnaird JA, Hutchinson D, Schurmann L, Nex PAM, de Lange R (2005) Petrology and mineralization of the southern Platreef: Northern limb of the Bushveld Complex, South Africa. Mineral Deposita 40:576–597
Kinnaird JA, Yudovskaya M, Naldrett AJ, Botha MJ, Chunnett GK (2012) PGE mineralisation in the Main Zone of the northern limb of the Bushveld Complex. In: 12th International Ni–Cu–(PGE) Symposium, Guiyang, China. Abstracts pp 73–76
Lambert DD, Walker RJ, Morgan JW, Shirey SB, Carlson RW, Zientek ML, Lipin BR, Koski MS, Cooper RL (1994) Re–Os and Sm–Nd Isotope Geochemistry of the Stillwater Complex, Montana: Implications for the Petrogenesis of the J-M Reef. J Petrol 35:1717–1753
Lee CA (1996) A review of mineralization in the Bushveld Complex and some other layered mafic intrusions. In: Cawthorn RG (ed) Layered intrusions. Elsevier Science, Amsterdam, pp 103–146
Lesher CM, Groves DG (1986) Controls on the formation of komatiite-associated nickel–copper sulphide deposits. In: Friedrich GH et al (eds) Geology and Metallogeny of Copper Deposits. Springer, Berlin, pp 43–62
Li C, Ripley EM (2005) Empirical equations to predict the sulfur content of mafic magmas at sulfide saturation and applications to magmatic sulfide deposits. Mineral Deposita 40:218–230
Li C, Barnes S-J, Makovicky E, Rose-Hansen J, Makovicky M (1996) Partitioning of nickel, copper, iridium, rhenium, platinum, and palladium between monosulfide solid solution and sulfide liquid: effects of composition and temperature. Geochim Cosmochim Acta 60:1231–1996
Lodders K (2003) Solar system abundances and condensation temperatures of the elements. Astrophys J 591:1220–1247
Lombard K (2012) Exploration results and mineral resource estimate for the Waterberg Platinum Project. Coffey Mining, South Africa, 89
Maier WD, Barnes S-J (1999) Platinum-group elements in silicate rocks of the Lower, Critical, and Main zones at Union section, western Bushveld Complex. J Petrol 40:1647–1671
Maier WD, Barnes S-J (2010) The petrogenesis of platinum-group element reefs in the Upper Main Zone of the Northern Lobe of the Bushveld Complex on the farm Moorddrift, South Africa. Econ Geol 105:841–854
Maier WD, de Klerk L, Blaine J, Manyeruke T, Barnes S-J, Stevens MVA, Mavrogenes JA (2008) Petrogenesis of contact-style PGE mineralization in the northern lobe in the Bushveld Complex: comparison of data from the farms Rooipoort, Townlands, Drenthe and Nonnenweth. Mineral Deposita 43:255–280
Mainwaring PR, Naldrett AJ (1977) Country-rock assimilation and the genesis of Cu–No sulfides in the Waterhen intrusion, Duluth Complex, Minnesota. Econ Geol 72:1269–1284
Manyeruke TD (2003) The petrology and geochemistry of the Platreef on the farms Townlands, near Potgietersrus, northern Bushveld Complex. M.Sc. thesis, Pretoria, South Africa, The University of Pretoria
Manyeruke TD, Maier WD, Barnes S-J (2005) Major and trace element geochemistry of the Platreef on the farm Townlands, northern Bushveld Complex. S Afr J Geol 108:381–396
McCallum IS (1996) The Stillwater Complex. In: Cawthorn RG (ed) Layered intrusions. Elsevier Science, Amsterdam, pp 441–484
McCutcheon SC, Kinnaird JA (2011) Platinum-group mineral assemblages in the Platreef at Tweefontein. Platreef Workshop, 4th, Mokopane, South Africa, 14th–16th January 2011, Abstracts
McDonald I, Harmer RE (2011) Cu–Ni–PGE mineralisation at Aurora is not Platreef, so where does it fit in? Platreef Workshop, 4th, Mokopane, South Africa, 14th-16th January 2011, Abstracts
McDonald I, Holwell DA (2007) Did lower zone magma conduits store PGE-rich sulphides that were later supplied to the Platreef? S Afr J Geol 110:611–616
McDonald I, Holwell DA (2011) Geology of the northern Bushveld Complex and the setting and genesis of the Platreef Ni–Cu–PGE deposit. Rev Econ Geol 17:297–327
McDonald I, Viljoen KS (2006) Platinum-group element geochemistry of mantle eclogites: a reconnaissance study of xenoliths from Orapa kimberlite, Botswana. Appl Earth Sci (Trans Inst Min Metall B) 115:81–93
McDonald AM, Cabri LJ, Stanley CJ, Rudashevsky NS, Poirier G, Mungall JE, Ross KC, Durham BR, Rudashevsky VN (2005a) Ungavaite, Pd4Sb3, a new intermetallic mineral species from the Mesamax Northwest deposit, Ungava region, Quebec, Canada: description and genetic implications. Can Mineral 43:1735–1744
McDonald I, Holwell DA, Armitage PEB (2005b) Geochemistry and mineralogy of the Platreef and ‘Critical Zone’ of the northern lobe of the Bushveld Complex, South Africa: implications for Bushveld stratigraphy and the development of PGE mineralization. Mineral Deposita 40:526–549
McDonald I, Holwell DA, Wesley B (2009) Assessing the potential involvement of an early magma staging chamber in the generation of the Platreef Ni–Cu–PGE deposit in the northern limb of the Bushveld Complex: a pilot study of the Lower Zone Complex at Zwartfontein. Appl Earth Sci (Trans Inst Min Metall B) 118:5–20
McDonald I, Jones RE, Holwell DA, Butler IB (2012) Platinum-group element tenors and S/Se ratios of Platreef sulphide melt inclusions. Platreef Workshop, 5th, Mokopane, South Africa, 9th–12th January 2012, Abstracts
Mountain BW, Wood SA (1988) Chemical controls on the solubility, transport, and deposition of platinum and palladium in hydrothermal solutions: a thermodynamic approach. Econ Geol 83:492–510
Mungall JE, Andrews DRA, Cabri LJ, Sylvester PJ, Tubrett M (2005) Partitioning of Cu, Ni, Au, and platinum-group elements between monosulfide solid solution and sulfide melt under oxygen and sulfur fugacities. Geochim Cosmochim Acta 69:4349–4360
Oberthür T, Cabri LJ, Weiser TW, McMahon G, Müller P (1997) Pt, Pd and other trace elements in sulfides of the main sulphide zone, great dyke, Zimbabwe: a reconnaissance study. Can Mineral 35:597–609
Penniston-Dorland SC, Wing BA, Nex PAM, Kinnaird JA, Farquhar J, Brown M, Sharman ER (2008) Multiple sulfur isotopes reveal a magmatic origin for the Platreef platinum group element deposit, Bushveld Complex, South Africa. Geology 36:979–982
Peregoedova AV (1998) The experimental study of the Pt–Pd–partitioning between monosulfide solid solution and Cu–Ni–sulfide melt at 900–840 °C. In: 8th International Platinum Symposium abstracts. Geological Society of South Africa and South African Institute of Minerals and Metallurgy, Symposium Series S18:325–373
Piña R, Gervilla F, Barnes S-J, Ortega L, Lunar R (2012) Distribution of platinum-group and chalcophile elements in the Aguablanca Ni–Cu sulfide deposit (SW Spain): evidence from a LA-ICP-MS study. Chem Geol 302–303:61–75
Piña R, Gervilla F, Barnes S-J, Ortega L, Lunar R (2013) Platinum-group elements-bearing pyrite from the Aguablanca Ni–Cu sulfide deposit (SW Spain): a LA-ICP-MS study. Eur J Mineral 25–2:241–252
Prichard HM, Barnes S-J, Maier WD, Fisher PC (2004) Variations in the nature of the platinum-group minerals in a cross-section through the Merensky Reef at Impala Platinum: implications for the mode of formation of the reef. Can Mineral 42:423–437
Prichard HM, Knight RD, Fisher PC, McDonald I, Zhou M-F, Wang CY (2013) Distribution of platinum-group elements in magmatic and altered ores in the Jinchuan intrusion, China: an example of selenium remobilization by post magmatic fluids. Mineral Deposita 48:767–786
Pronost J, Harris C, Pin C (2008) Relationship between footwall composition, crustal contamination, and fluid–rock interaction in the Platreef, Bushveld Complex, South Africa. Mineral Deposita 43:825–848
Ripley EM (1981) Sulfur isotopic abundances of the Dunka Road Cu–Ni deposit, Duluth Complex, Minnesota. Econ Geol 76:619–620
Ripley EM, Lambert DD, Frick LR (1986) Re-Os, Sm-Nd, and Pd isotopic constraints on mantle and crustal contributions to magmatic sulfide mineralization in the Duluth Complex. Geochim Cosmochim Acta 62:3349–3365
Sharman ER, Penniston-Dorland SC, Kinnaird JA, Nex PAM, Brown M, Wing BA (2013) Primary origin of marginal Ni–Cu–(PGE) mineralization in layered intrusions: Δ33 S evidence from the Platreef, Bushveld, South Africa. Econ Geol 108:365–377
Sharman-Harris E, Kinnaird JA, Harris C, Horstmann UE, Wing B (2005) A new look at sulfide mineralization of the northern limb, Bushveld Complex: a stable isotope study. Appl Earth Sci (Trans Inst Min Metall B) 114:B252–B263
Smith JW, Holwell DA, McDonald I (2011) The mineralogy and petrology of platinum-group element-bearing sulfide mineralization within the Grasvally Norite–Pyroxenite–Anorthosite (GNPA) member, south of Mokopane, northern Bushveld Complex, South Africa. Appl Earth Sci (Trans Inst Min Metall B) 120:B158–B174
Smith JW, Holwell DA, McDonald I (2012) An overview of PGE and BMS mineralization within the GNPA member. Platreef Workshop, 5th, Mokopane, South Africa, 9th–11th November 2012, Abstracts
Smith JW, Holwell DA, McDonald I, Boyce AJ (2013) The emplacement and relative timing of PGE mineralisation within the northern Bushveld Complex. In: Jonsson E et al. (eds) Mineral deposit research for a high-tech world. Proceedings of the 12th Biennial Meeting, Uppsala, Sweden 2:1061–1064
Sutherland A-J (2013) The nature and genesis of PGE-rich sulphide horizons on the farm War Springs, northern Bushveld Complex, South Africa. MGeol thesis, England, University of Leicester
Tomkins AG (2010) Wetting facilitates late-stage segregation of precious metal-enriched sulfosalt melt in magmatic sulfide systems. Geology 38:951–954
Twist D (1985) Geochemical evolution of the Rooiberg silicic lavas in the Loskop Dam area; Southeastern Bushveld. Econ Geol 80:1153–1165
van der Merwe MJ (1976) The layered sequence of the Potgietersrus limb of the Bushveld Complex. Econ Geol 71:1337–1351
van der Merwe MJ (1978) The geology of the basic and ultramafic rocks of the Potgietersrus limb of the Busveld Complex. Ph.D. thesis, South Africa, University of the Witwatersrand
van der Merwe MJ (2008) The geology and structure of the Rustenburg Layered Suite in the Potgietersrus/Mokopane area of the Bushveld Complex, South Africa. Mineral Deposita 43:405–419
von Gruenewaldt G, Hulbert LJ, Naldrett AJ (1989) Contrasting platinum group element concentration patterns in cumulates of the Bushveld Complex. Mineral Deposita 24:219–229
Walraven F (1985) Genetic aspects of the granophyric rocks of the Bushveld Complex. Econ Geol 80:1166–1180
Walraven F, Hattingh E (1993) Geochronology of the Nebo Granite Bushveld Complex. S Afr J Geol 96:31–41
Wilson A, Chunnett G (2006) Trace element and platinum group element distributions and the genesis of the Merensky Reef, Western Bushveld Complex, South Africa. J Petrol 47:2369–2403
Wood SA (2002) The aqueous geochemistry of the platinum-group elements with applications to ore deposits. In: Cabri LJ (ed) The geology, geochemistry, mineralogy and mineral beneficiation of platinum-group elements. Canadian Institute of Mining, Metallurgy and Petroleum, pp 211–249
Acknowledgments
The authors would like to thank Caledonia Mining Corporation and in particular Trevor Pearton, for allowing access to the drillcore on the farms Rooipoort, Grasvally, Moorddrift and War Springs, and giving permission to publish this work. Jennifer Smith’s Ph.D. research is funded by the Natural Environment Research Council (NE/1528426/1). Louis Cabri and an anonymous reviewer are thanked for their constructive comments on improving the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: B. Lehmann
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Smith, J.W., Holwell, D.A. & McDonald, I. Precious and base metal geochemistry and mineralogy of the Grasvally Norite–Pyroxenite–Anorthosite (GNPA) member, northern Bushveld Complex, South Africa: implications for a multistage emplacement. Miner Deposita 49, 667–692 (2014). https://doi.org/10.1007/s00126-014-0515-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-014-0515-6