
Abstract
Aims/hypothesis
Accelerated migration and proliferation of vascular smooth muscle cells (VSMCs) enhances arterial restenosis after angioplasty in insulin resistance and diabetes. Elevation of Src homology 2-containing protein tyrosine phosphatase 1 (SHP-1) induces apoptosis in the microvasculature. However, the role of SHP-1 in intimal hyperplasia and restenosis has not been clarified in insulin resistance and diabetes.
Methods
We used a femoral artery wire injury mouse model, rodent models with insulin resistance and diabetes, and patients with type 2 diabetes. Further, we modulated SHP-1 expression using a transgenic mouse that overexpresses SHP-1 in VSMCs (Shp-1-Tg). SHP-1 agonists were also employed to study the molecular mechanisms underlying the regulation of SHP-1 by oxidised lipids.
Results
Mice fed a high-fat diet (HFD) exhibited increased femoral artery intimal hyperplasia and decreased arterial SHP-1 expression compared with mice fed a regular diet. Arterial SHP-1 expression was also decreased in Zucker fatty rats, Zucker diabetic fatty rats and in patients with type 2 diabetes. In primary cultured VSMCs, oxidised LDL suppressed SHP-1 expression by activating Mek-1 (also known as Map2k1) and increased DNA methylation of the Shp-1 promoter. VSMCs from Shp-1-Tg mice exhibited impaired platelet-derived growth factor (PDGF)-stimulated tyrosine phosphorylation with a concomitant decrease in PDGF-stimulated VSMC proliferation and migration. Similarly, HFD-fed Shp-1-Tg mice and mice treated with the SHP-1 inducer, Icariside II, were protected from the development of intimal hyperplasia following wire injury.
Conclusions/interpretation
Suppression of SHP-1 by oxidised lipids may contribute to the excessive VSMC proliferation, inflammatory cytokine production and intimal hyperplasia observed in arteries from diabetes and insulin resistance. Augmenting SHP-1 levels is a potential therapeutic strategy to maintain stent patency in patients with insulin resistance and diabetes.
Similar content being viewed by others


Introduction
A prominent feature in the pathology of restenosis and atherosclerosis is the increased number of vascular smooth muscle cells (VSMCs) in the intima of arteries due to enhanced VSMC migration and proliferation [1, 2]. The key role of abnormal VSMC growth in the pathogenesis of arterial restenosis is highlighted by the three- to fourfold reduction in restenosis rates with the use of drug-eluting stents that target smooth muscle cell proliferation and migration [3, 4]. However, even with the use of these stents, patients with diabetes still exhibit an increased risk of restenosis compared with non-diabetic patients [5].
Multiple mechanisms have been proposed to explain the excessive VSMC proliferation observed in diabetes and other insulin-resistant states. Hyperglycaemia and dyslipidaemia are thought to play important roles by increasing the expression of mitogenic growth factors [6]. Indeed, elevated expression of insulin, insulin-like growth factor-1 (IGF-1), platelet-derived growth factor (PDGF) and fibroblast growth factor-2 (FGF2) [7–9] has been associated with enhanced VSMC proliferation. However, there is disagreement in the literature about whether the expression of these growth factors is increased in the vascular wall or plasma in the context of diabetes [10]. Thus, it is still unclear if this mechanism drives excessive VSMC proliferation in the diabetic milieu.
In contrast, the possibility of enhanced action of growth factors through amplification of their intracellular signalling cascades has not been studied in detail. Src homology-2-containing protein tyrosine phosphatase (SHP-1) is a tyrosine phosphatase that functions as a negative regulator of growth-factor-dependent signalling. SHP-1 is a potent negative regulator of growth-factor signalling but the expression of SHP-1 and its contribution to vascular restenosis in animal models of obesity and type 2 diabetes have not been investigated.
In this report, we aimed to determine the expression level of SHP-1 in the arterial wall in animal models and human obesity and type 2 diabetes mellitus. Further, we studied the effect of oxidised lipids on SHP-1 expression in VSMCs and contribution to the excessive VSMC proliferation, inflammatory cytokine production and intimal hyperplasia observed in arteries leading to a high risk of restenosis in insulin resistance and diabetes. Finally, we aimed to determine whether pharmacologically augmenting SHP-1 expression could be an effective therapeutic approach to reduce restenosis.
Methods
For detailed methods, please refer to the electronic supplementary materials (ESM) Methods.
Animals and reagent
All protocols for animal use and euthanasia were reviewed and approved by the Animal Care Committee of the Joslin Diabetes Center. The experiments were in accordance with National Institutes of Health (NIH) guidelines following the standards established by the Animal Welfare Acts and by the documents entitled ‘Principles for Use of Animals’ and ‘Guide for the Care and Use of Laboratory Animals’. Male Zucker lean (ZL) rats, Zucker fatty (ZF) rats, Zucker diabetic fatty (ZDF) and their control (LEAN +/?) were from Charles River (Wilmington, MA, USA). C57BL/6 mice were from Jackson Laboratory (Bar Harbor, ME, USA). PDGF-BB was purchased from Sigma (St Louis, MO, USA).
Procurement of human arteries
This work was approved by the regional ethics committee (Region Syddanmark, protocol number ID S-20100044). All patients over the age of 18 years, referred to coronary artery bypass grafting (CABG) surgery at Odense University Hospital, Denmark were asked to participate. Type 2 diabetes was defined as a history of diabetes, treatment with glucose-lowering medicine or HbA1c > 6.5% and absence of IA2 and GAD65 autoantibodies. Arteries from a part of the internal thoracic artery were collected. Perivascular tissue was removed and arterial rings were fixed in formalin for paraffin embedding.
Generation of SM22α-promoter-driven Shp-1-overexpressing transgenic mouse line
CMV promoter drives 1.57 kb Lox-stop-Lox (LSL) fragment followed by full-length Shp-1 (also known as Ptpn6) cDNA cloned into Not1 and Xho1 (Fig. 3a). The final construct was digested with BstBI and Mlu and the 6.5 kb fragment was injected into blastocytes. LSL-Shp-1 mice were crossbred with Sm22α-CreKI mice (stock no. 006878, Jackson Laboratory) to generate mice overexpressing Shp-1 specifically in VSMCs, maintained on C57BL/6J background. Primers (see ESM Methods for details) were used for LSL-Shp-1 genotyping.
Femoral artery wire injury and histological analysis
Bilateral wire injury of the femoral artery was performed as previously described [11]. Mice were sacrificed on day 28 post arterial injury. Arteries were excised and paraffin-embedded. Sections (5 μm) were stained using Verhoeff tissue elastin stain (Sigma) and were masked for the measurement of the luminal, intimal and medial areas using NIH ImageJ (Bethesda, MD, USA).
Mouse aortic smooth muscle cell culture
Aortas were dissected and digested with collagen II (Worthington, Columbia, NJ, USA). The adventitia was removed and further digested and grown in 20% DMEM. Cells at passage 2–3 and that had been starved for 48 h were used for all the experiments.
VSMC transfection
Small interfering RNA for scramble or Mek1 (also known as Map2k1) (sc-29396; Santa Cruz, Dallas, TX, USA) and Jnk (also known as Mapk8) (no. 6232; Cell Signaling Beverly, MA, USA) were transfected at 10 nmol/l using Basic Smooth Muscle Cells (SMC) Nucleofector Kit.
Methylation analysis of the Shp-1 promoter by bisulfite sequencing
Genomic DNA was extracted from VSMCs and bisulfite conversion was performed using EZ DNA Methylation Gold kit (Zymo Research, Irvine, CA, USA). The CT-converted DNA was then amplified (see ESM Methods for primer details). The PCR fragment was cloned into a TOPO vector for DNA sequencing. Methylation was analysed using BiQ Analyzer software (Max-Planck-Institut für Informatik, Saarbrücken, Germany).
SHP-1 activity assay
SHP-1 was immunoprecipitated with 2 μg anti-SHP-1 polyclonal antibody (Santa Cruz) from control and Shp-1-Tg VSMCs overnight, 20 μl Protein A/G PLUS-Agarose (Santa Cruz) was added and beads were washed and then assayed by RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (Molecular Probes, Eugene, OR, USA).
Cell proliferation assays, cell cycle analysis and migration assays
Cell proliferation was measured using Click-iT EdU Flow Cytometry Assay Kit (Invitrogen, Carlsbad, CA, USA). The CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT assay; Promega, Madison, WI, USA) was used for the measurement of cell number. For cell cycle analysis, cells were and digested with RNase A. Propidium iodide was added before flow cytometry analysis. Migration was measured by Transwell insert using CytoSelect 24-Well Cell Migration Assay kit (Cell Biolabs, San Diego, CA, USA).
Quantitative real-time PCR (qPCR) analysis
Total RNA was isolated and purified and converted into cDNA. Shp-1, Shp-2 (also known as Ptp11), Ptp1b (Ptpn1), Cyclin E1 (Ccne1), Mcp-1 (Ccl2) and 36B4 (Rplp0). qPCR primers were used with SYBR Green Master mix kit and detection was performed using the ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Relative change was determined using the ΔΔ method and normalised to 36B4 (also known as Rplp0).
Western blotting
Protein samples were electrophoresed in Bio-Rad TGX precast gels, transferred to a nitrocellulose membrane and incubated with primary antibodies (see ESM Methods for details). For p-Akt and Akt, and p-ERK and ERK (same molecular mass protein), two separate gels were used for phosphorylated and unphosphorylated protein instead of stripping the membrane to reprobe the other to avoid the inefficiency of stripping. Quantitative densitometry was performed using NIH Image J.
Immunofluorescent staining
Mouse femoral arteries were fixed, embedded in paraffin and 5 μm sections were stained with antibodies (see ESM Methods for details). Photoshop (San Jose, CA, USA) was used for the quantification of SHP-1 expression levels in the media of arteries. For the Ki-67 and macrophage (MAC2) staining, positive cells with the overlay of Ki-67 or MAC2 (red) with smooth muscle cell marker (green) and DAPI (nucleus) were counted.
Icariside II oral gavaging in HFD-fed mice
Icariside II (Syd Lab, Natick, MA, USA) was dissolved in 15% PEG 400 (Sigma) and given to mice fed a high-fat diet (HFD) via daily oral gavaging. 15% PEG 400 was given to vehicle-treated mice. Treatment started 1 day before femoral artery injury.
Statistics
Comparison of two groups was made using unpaired t test. Comparison of more than two groups was performed by one-way ANOVA followed by the post hoc analysis with unpaired t test to evaluate statistical significance between the two groups. The data are presented as means ± SEM, unless otherwise stated. Statistical significance was defined as p < 0.05.
Results
HFD feeding enhances femoral artery intimal hyperplasia in response to wire injury
To determine whether insulin resistance and diabetes increased intimal hyperplasia, wire injury in the femoral artery was performed on mice fed either a regular diet (RD) or an HFD (60% energy from fat) for 4 weeks starting from the age of 8 weeks. HFD-fed mice exhibited hypercholesterolaemia, obesity and insulin resistance as determined by measurement of plasma lipids and insulin levels and intraperitoneal insulin tolerance (IP-ITT) and glucose tolerance test (IP-GTT) (ESM Fig. 1a–c and ESM Table 1). Intimal hyperplasia in response to wire injury showed that the intimal area and intima/media ratio were increased by 100% and 133%, respectively, in HFD-fed mice compared with RD-fed mice (Fig. 1a).

Insulin resistance and diabetes induce severe intimal hyperplasia in response to arterial injury and downregulate SHP-1 expression in rodent aortas and human arteries. (a) C57BL/6J mice were fed either RD or HFD for 4 weeks before injury was performed; n = 5–6; scale bar, 50 μm. (b) C57BL/6J mice were fed on either RD or HFD for 8 weeks; n = 7. (c, d) Aortas from ZF and ZDF rats at 14 weeks of age; n = 5. (e) Human mammary arteries; scale bar, 50 μm; n = 6. AU, arbitrary units; T2DM, type 2 diabetes. *p < 0.05 and **p < 0.01 for indicated comparison
Insulin resistance and diabetes downregulates SHP-1 expression in rodent aorta and human arteries
Expression of SHP-1 in the aorta was measured in three models of obesity, insulin resistance and type 2 diabetes, including mice with diet-induced obesity, ZF rats and ZDF rats. SHP-1 protein was significantly decreased in aortas from HFD-fed mice, ZF rats and ZDF rats compared with their respective controls (Fig. 1b–d). In contrast, expression of other protein tyrosine phosphatases (PTPs) PTP1B and SHP-2 was not changed in the femoral arteries from HFD vs. RD mice or in the neointima and media of injured femoral artery (ESM Fig. 1d, e). Thus, SHP-1, but not other PTPs, is decreased in insulin resistance and diabetes. SHP-1 protein levels were also significantly decreased in the media of mammary arteries from patients with type 2 diabetes compared with non-diabetic patients (Fig. 1e) as assessed by double immunostaining of SHP-1 and α-smooth muscle actin (α-SMA). Clinical characteristics of the patients with type 2 diabetes included elevated levels of HbA1c, total cholesterol, LDL and triacylglycerol levels when compared with individuals without type 2 diabetes (ESM Table 2).
Effect of lipids and glucose on SHP-1 expression and its mechanisms
As HFD mice and ZF and ZDF rats exhibited hyperlipidaemia, hyperglycaemia and hyperinsulinemia (ESM Table 1 and ESM Fig. 1a–c), we evaluated the effects of modified lipids and hyperglycaemia on the regulation of SHP-1 expression in mouse aortic VSMCs in vitro. In primary cultures of VSMCs from C57BL/6J mice, the expression of Shp-1 mRNA (Fig. 2a) and protein (Fig. 2b) was decreased by oxidised LDL (oxLDL) and acetyl-LDL (acLDL). No change in SHP-1 expression was observed when VSMCs were incubated in high (25 mmol/l) vs. low (5.5 mmol/l) glucose conditions (Fig. 2a, b) or were treated with insulin (ESM Fig. 2a, b). In the presence of actinomycin D, the half-life of Shp-1 mRNA in VSMCs was 4 h and was not changed by treatment with oxLDL (Fig. 2c). Since oxLDL activates several stress kinases, we studied the effect of oxLDL on the activation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) (MEK), p38MAPK and c-Jun N-terminal kinase (JNK). All three were activated by oxLDL (Fig. 2d). However, only silencing Mek1 impaired the ability of oxLDL to suppress SHP-1 expression (Fig. 2e, f and ESM Fig. 3a); we did not observe any change in VSMCs with p38αMAPK, p38βMAPK or JNK knockdown (ESM Fig. 3b–e). Prior study has suggested downregulation of SHP-1 expression could be mediated by increased DNA methylation of its promoter in cancer cells [12]. To delineate the mechanism underlying regulation of SHP-1 by oxLDL, we studied the effect of oxLDL treatment on methylation at the Shp-1 promoter. Bisulfite sequencing was performed on the region of the Shp-1 promoter ranging between +593 and +879 relative to the +1 transcription start site of the Shp-1 gene. We observed a 68% increase in unconverted cytosines on the Shp-1 promoter in VSMCs treated with oxLDL vs control (Fig. 2g, h), suggesting that DNA methylation was increased in the Shp-1 promoter. Thus, the expression of SHP-1 is decreased in diabetes potentially via oxLDL induced-methylation of the Shp-1 promoter.

oxLDL decreases SHP-1 expression in VSMCs via elevating DNA methylation on the Shp-1 promoter. (a, b) VSMCs were starved for 24 h in 0.1% fatty-acid-free BSA control, or treated with 100 μg/ml oxLDL (ox), 100 μg/ml acLDL (ac) or 25 mmol/l d-glucose (HG) for 8 h. Shp-1 mRNA (a) and protein (b) were analysed; n = 3 and 5, respectively. (c) VSMCs were treated with 5 μg/ml actinomycin with or without oxLDL; n = 3; squares, 0.1% BSA; circles, oxLDL. (d) VSMCs treated with control and oxLDL; n = 3. (e) VSMCs transfected with 10 nmol/l scramble (siControl) or Mek1 small interfering RNA (siMek1) and treated with oxLDL. (f) VSMCs treated with control and oxLDL in the presence or absence of 10 μmol/l PD98059 (PD) for 24 h; n = 5. (g, h) Bisulfite sequencing at the region of Shp-1 promoter. Data are presented as the ratio of unconverted cytosine (5-methylcytosine, 5meC) and total cytosines. *p < 0.05, **p < 0.01 and ***p < 0.001 vs control or for indicated comparison
Generation and characterisation of Shp-1-Tg mice
To support the idea that enhancing SHP-1 expression in VSMCs can decrease intimal hyperplasia in insulin resistance and type 2 diabetes, we generated transgenic mice overexpressing SHP-1 specifically in VSMCs (Fig. 3a). Shp-1 mRNA and protein levels were increased more than twofold in aortas and VSMCs of Shp-1-Tg mice compared with control mice (Fig. 3b–d). Further, SHP-1 activity was increased twofold in VSMCs from Shp-1-Tg mice compared with control mice (Fig. 3e). In contrast, mRNA expression of other PTPs, Ptp1b and Shp-2, was not changed in VSMCs from Shp-1-Tg mice vs. control mice (ESM Fig. 4a).

Generation and characterisation of Shp-1-Tg mice. (a) Schema of LSL-Shp-1 mice crossbred with Sm22α-CreKI mice to generate mice overexpressing ShP-1 specifically in VSMCs. (b–e) SHP-1 mRNA and protein from aortas and aortic SMCs from control and Shp-1-Tg mice; n = 3–5. Tg, Shp-1-Tg mice. *p < 0.05, **p < 0.01 and ***p < 0.001 for indicated comparison
SHP-1 overexpression inhibits the activation of PDGF, insulin and FGF2 receptors and their downstream signalling in VSMCs
The mitogenic actions of growth factors such as PDGF, FGF2 and insulin can affect VSMC migration and proliferation [13–15]. These factors signal via receptor tyrosine kinases and their actions may be suppressed by SHP-1. Thus, we determined whether overexpression of SHP-1 inhibited the activation of tyrosine kinase receptors. First, in VSMCs isolated from Shp-1-Tg mice tyrosine phosphorylation of PDGF receptor- β (PDGFRβ) was significantly decreased in response to PDGF-BB (Fig. 4a). We then determined whether SHP-1 targets tyrosine phosphorylation at a specific site of on PDGFRβ. Three tyrosine phosphorylation sites (Tyr751, Tyr740, Tyr771) were reported to be stimulated by PDGF-BB [16]. Tyr751 was significantly decreased in VSMCs from Shp-1-Tg mice vs. control mice in response to PDGF-BB (Fig. 4b). No difference was observed at Tyr740 or Tyr771 (ESM Fig. 4b,c). Thus, SHP-1 inhibits the action of PDGF-BB via targeting Tyr751. Activation of p-ERK by PDGF-BB (0.1 and 0.5 ng/ml) was decreased significantly in VSMCs from Shp-1-Tg mice (Fig. 4c, d). Interestingly, no difference was observed in Akt activation (p-Akt) between control mouse and Shp-1-Tg mouse VSMCs at basal or with PDGF stimulation (Fig. 4e).

SHP-1 overexpression inhibits the activation of PDGFRβ and FGF2 receptors and their downstream signalling in VSMCs. (a) VSMCs stimulated with 2.5 ng/ml PDGF-BB for 5 min. n = 4 per group. (b) Tyr751 stimulated with PDGF-BB for 5 min; n = 4. (c–e) VSMCs stimulated with PDGF-BB for 5 min; n = 4. (f–h) VSMCs stimulated with FGF2 for 5 min. C, VSMCs from control mice; T, VSMCs from Shp-1-Tg mice; IB, immunoblot; IP, immunoprecipitation; n = 3. *p < 0.05 and ***p < 0.001 for indicated comparisons
FGF2 signalling in VSMCs was also studied because activated platelets secrete FGF2, resulting in VSMC migration and proliferation during restenosis [14]. Both p-ERK and p-Akt levels induced by FGF2 were significantly decreased in VSMCs from Shp-1-Tg mice (Fig. 4f–h). Moreover, overexpression of SHP-1 also decreased insulin receptor β tyrosine phosphorylation and p-ERK activation in response to insulin but insulin-stimulated increases in p-Akt were not affected (ESM Fig. 4d–f).
Overexpression of SHP-1 in VSMCs inhibits the activation of ERK and Akt in vivo
The effect of overexpressing SHP-1 on p-ERK and p-Akt activation in vivo was studied in control mice and Shp-1-Tg mice fed with HFD for 8 weeks before femoral artery wire injury. No significant differences in body weight, blood pressure, glucose tolerance, insulin tolerance or plasma lipid concentrations between control and Shp-1-Tg mice on HFD were observed (ESM Fig. 5a–g). Further, we did not observe any difference in Shp-1 mRNA expression between the sham and injury groups (ESM Fig. 6).
After 8 weeks on HFD, basal levels of p-ERK were similar in control and Shp-1-Tg mice with sham injury (Fig. 5a, b). In contrast, wire injury increased p-ERK expression in the femoral artery twofold compared with control mice with sham injury; this effect was significantly decreased in injured femoral arteries from Shp-1-Tg mice (Fig. 5b). In addition, p-Akt activation in Shp-1-Tg and control mice on HFD after wire injury showed a fourfold increase; this was significantly inhibited in Shp-1-Tg mice (Fig. 5c). Similarly, PDGF signalling was enhanced in whole aortas from Shp-1-Tg mice stimulated with PDGF-BB ex vivo (Fig. 5d–f).

Overexpression of SHP-1 in VSMCs inhibits the activation of ERK and Akt in vivo in response to arterial injury in mice fed an HFD. (a–c) Mice on HFD for 8 weeks at 1 h post injury. (d–f) Aortas incubated in 100 ng/ml PDGF-BB for 10 min. C, control mice, T, Shp-1-Tg mice. n = 5. *p < 0.05, **p < 0.01 and ***p < 0.001 for indicated comparisons
Inhibition of cell proliferation, cell cycle progression and migration in VSMCs from Shp-1-Tg mice
PDGF-BB increased cell proliferation in VSMCs from control mice as measured by BrdU incorporation and MTT assay. This effect was reduced in VSMCs from Shp-1-Tg mice (Fig. 6a, b and ESM Fig. 7a). Cell cycle analysis showed that PDGF-BB-induced cell cycle progression from G1 to S phase was inhibited by SHP-1 overexpression in VSMCs (Fig. 6c). In keeping with these findings, PDGF-BB-induced cyclin E1 expression was decreased in VSMCs from Shp-1-Tg mice (Fig. 6d). In contrast, the rate of VSMC apoptosis in Shp-1-Tg mice was no different from that in control mice (ESM Fig. 7b). We also assessed whether overexpression of SHP-1 could affect VSMC migration in response to PDGF-BB. Migration assays showed that PDGF-induced migration was reduced significantly in VSMCs from Shp-1-Tg mice compared with control mice (Fig. 6e). Together, our findings demonstrate that SHP-1 overexpression impairs mitogenic signalling and limits growth-factor-induced proliferation and migration of VSMCs.

Overexpression of SHP-1 in VSMCs inhibits proliferation, cell cycle progression and migration. (a) VSMCs stimulated with or without 10 ng/ml PDGF-BB for 16 h; n = 4. (b) VSMCs with or without PDGF-BB (PDGF) for 48 h; n = 4. (c) VSMCs stimulated with or without PDGF-BB for 24 h; n = 5. C-PDGF, control VSMCs treated with PDGF. (d) Cyclin E1 mRNA; n = 4. (e) VSMCs stimulated with or without PDGF-BB for 8 h. Migrated cells measured by Transwell; n = 4. (f) VSMCs stimulated with or without 10 ng/ml FGF2 or PDGF-BB for 8 h; n = 3. (g) VSMCs pre-treated with 50 μmol/l PD98059 or 100 nmol/l wortmannin for 30 min and co-incubated with FGF2 or PDGF-BB for 8 h; n = 4. C, control VSMCs; T, Shp-1-Tg VSMCs. EdU, ethynyldeoxyuridine; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; *p < 0.05, **p < 0.01 and ***p < 0.001 for indicated comparisons (or vs no treatment in g); † p < 0.05 vs PDGF-BB vehicle or FGF2 vehicle
Overexpression of SHP-1 in VSMCs inhibits the expression of inflammatory cytokine monocyte chemoattractant protein-1
The number of inflammatory cells in the arterial wall after injury may contribute to the severity of the restenosis process [17]. Therefore, we characterised the effect of PDGF-BB and FGF2 on the expression of monocyte chemoattractant protein-1 (MCP-1) in VSMCs. FGF2 and PDGF-BB increased Mcp-1 mRNA levels in VSMCs from control mice but this induction was relatively impaired in VSMCs from Shp-1-Tg mice (Fig. 6f). Mechanistically, a selective inhibitor of the p-ERK pathway, PD98059, significantly inhibited the effect of FGF2 and PDGF-BB on Mcp-1 mRNA expression. The addition of wortmannin, an inhibitor of the phosphoinositide 3-kinase/Akt pathway, impaired the ability of PDGF-BB to induce MCP-1 expression but had no effect on FGF2-dependent increases in MCP-1 (Fig. 6g).
Reduction of intimal hyperplasia in response to injury in HFD-fed Shp-1-Tg mice
To determine further the causal relationship between decreased SHP-1 expression and accelerated intimal hyperplasia in insulin resistance and diabetes, we studied the severity of intimal hyperplasia in response to wire injury in the femoral artery from Shp-1-Tg and control mice after 8 weeks of HFD feeding. The intimal area measured at 28 days after injury was reduced by 43% in Shp-1-Tg mice compared with control mice. The ratio of intimal to medial area also exhibited a reduction of 54% in Shp-1-Tg compared with control mice on HFD (Fig. 7a).

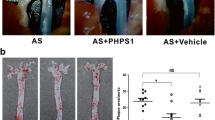
HFD-fed mice overexpressing SHP-1 develop less intimal hyperplasia and exhibit a decrease in cell proliferation and macrophage content in the vascular wall lesions in response to injury. (a) Mice were fed HFD for 8 weeks and examined at day 28 post injury; n = 7–10, scale bar, 100 μm. (b–e) Mice were fed HFD for 8 weeks and examined at day 7 post injury. The number of positive Ki-67 and αSMA were counted; n = 8, scale bar, 20 μm (b, c). The number of positive MAC2 in the media was counted; n = 5 arteries per group; scale bar, 20 μm (d, e). (f, g) Mice were fed HFD for 8 weeks and 100 mg/kg Icariside II was administered 1 day before injury via daily oral gavaging for 28 days. n = 4–6 arteries, scale bar, 50 μm (f). (g) Intimal area and intimal–medial ratio were quantified and analysed; n = 13–16, scale bar, 20 μm. *p < 0.05 and ***p < 0.001 for indicated comparisons
Cell proliferation and macrophage content in the vascular wall in response to injury
To confirm that the suppression of proliferation and pro-inflammatory cytokine expression observed in SHP-1-overexpressing VSMCs mediated the protective action of SHP-1 in arterial injury, we studied VSMC proliferation and macrophage number in the wall of the femoral artery 8 days after wire injury. Proliferating smooth muscle cells in the injured artery, as determined by staining α-SMA and Ki67, were counted in the media and neointima. Double-immunostained cells were significantly less abundant in Shp-1-Tg mice than in control mice (Fig. 7b, c). Furthermore, the macrophage content of media, as quantified by immunostaining for MAC2, was significantly decreased in the media of the femoral arteries from Shp-1-Tg mice compared with control mice after injury (Fig. 7d, e).
Reduction of intimal hyperplasia in response to injury in HFD mice treated with SHP-1 activator
Icariside II was reported to increase SHP-1 expression and have an antiproliferative effect in cancer cells [18]. Further, Icariside II exhibited a reasonable pharmacokinetics profile in a single dose oral gavaging study [19]. Thus, Icariside II was given 1 day before mice underwent arterial injury at a dose of 100 mg/kg via daily oral gavaging for 28 days. In line with previous studies [18], we observed that SHP-1 expression was increased in the femoral arteries of Icariside II-treated mice when compared with mice treated with vehicle (Fig. 7f). Notably, we observed significant reduction in intimal area and the ratio of intimal to medial area at 28 days after injury in HFD-fed mice treated with Icariside II when compared with HFD-fed mice treated with vehicle (Fig. 7g).
Discussion
The results from this study demonstrate that SHP-1 expression in arterial VSMCs is decreased in insulin-resistant and diabetic rodents and in patients with type 2 diabetes. In addition, overexpression of SHP-1 in VSMCs, induced by both genetic and pharmacological approaches, protected against intimal hyperplasia in response to arterial wire injury by inhibiting growth-factor-dependent signalling.
The mitogenic effects of diabetes and insulin resistance on VSMCs in both restenosis and atherosclerosis have always been puzzling considering the increases in apoptosis observed in many other vascular cells including retinal pericytes, podocytes and even endothelial cells [20–22]. Interestingly, we previously demonstrated that diabetes and hyperglycaemia upregulate the expression of SHP-1 in vascular contractile cells from the capillaries of the retina and glomeruli [20, 22]. However, in the current study hyperglycaemia had no effect on SHP-1 expression in arterial VSMCs, whereas modified lipids suppressed SHP-1 expression. Thus, these findings identify differential regulation of SHP-1 by hyperglycaemia and lipids in the micro- and macrovasculature as a potential mechanistic explanation for the dichotomous pathologies of contractile cells in macro- and microvessels in diabetes [20, 23].
Dyslipidaemia rather than dysglycaemia seems to drive the changes in SHP-1 expression in animal models of obesity and diabetes as in vitro studies demonstrated that treatment with modified lipids, but not hyperglycaemia suppressed SHP-1 expression. The pathogenic effects of hyperlipidaemia on cells are mainly mediated by modified LDL such as oxLDL and acLDL. We found that both oxLDL and acLDL inhibited SHP-1 expression indicating that decreased SHP-1 expression in hyperlipidaemic status is not oxLDL-specific and another modified LDL could also be involved. Further studies will be needed to confirm that these findings are relevant to human diseases. However, our finding, in a limited cohort, that SHP-1 expression is suppressed in type 2 diabetes patients is encouraging.
Overexpression of SHP-1 in VSMCs from RD-fed mice decreased the mitogenic action of several growth factors including PDGF-BB, FGF2 and insulin. SHP-1 overexpression exerts this action via inhibition of tyrosine phosphorylation at the receptor level via Tyr751 on PDGFRβ, thus inhibiting activation of p-ERK and preventing growth-factor-induced cell cycle progression. It has been reported that the phosphorylation of Tyr751 is required for both phosphoinositide 3-kinase and GTPase-activating protein (GAP) binding in epithelial cells [16, 24]. Further studies are needed to determine the mechanistic significance of the reduction in Tyr751 of PDGFRβ in VSMCs.
Our study is consistent with previous findings of the inhibitory effect of SHP-1 on the ERK pathway [25] and PDGFR signalling [26] in VSMCs. The activation of p-Akt in response to PDGF-BB and insulin stimulation of VSMCs was not affected by SHP-1 overexpression. In contrast, stimulation by PDGF in ex vivo experiments showed that the signalling of both p-Akt and p-ERK were diminished and that SHP-1 overexpression could significantly reduce p-Akt and p-ERK induction by arterial injury. It is possible that the in vivo effect of SHP-1 alters the action of multiple growth factors in the Akt pathway. Interestingly, potentiation of phosphatases upstream of Akt have previously been suggested as a therapeutic strategy in the context of vascular injury and restenosis [27]. Our work validates this suggestion in the context of diabetes and obesity. Elucidation of the role of regulatory phosphatases other than SHP-1 may allow us to fine tune the function of vascular mitogens and optimise growth-factor-dependent signalling in response to vascular injury [27–29].
Importantly, induction of SHP-1 overexpression in VSMCs using both genetic and pharmacological approaches was sufficient to normalise the response to wire injury in mice with diet-induced obesity. These results indicate that therapeutic intervention to induce gain of SHP-1 function or normalisation of SHP-1 levels in stents could decrease restenosis in diabetes and obesity. While SHP-1 is a potential therapeutic target, translational challenges exist. Kappert’s group have shown that systemic administration of an SHP-1 inhibitor improves insulin sensitivity in animal models of diet-induced obesity [30]. Moreover, HFD increases SHP-1 in a number of tissues [23]. Thus, future therapeutic strategies to activate SHP-1 and prevent vascular restenosis should consider targeted delivery of SHP-1 modulators either via topical application in the form of drug-eluting stents or cell-specific delivery using targeted delivery strategies such as ligand-conjugated nanoparticles [31].
In summary, our findings suggest a new perspective on how diabetes and insulin resistance increase the proliferation and migration of VSMCs. oxLDL-mediated downregulation of SHP-1 enhances growth-factor signalling in insulin resistance and diabetes and enhances intimal hyperplasia in response to arterial injury. These results support manipulation of SHP-1 activity or expression as a novel therapeutic approach to prevent restenosis and improve angioplasty outcomes in patients with insulin resistance and diabetes.
Abbreviations
- acLDL:
-
Acetyl-LDL
- ERK:
-
Extracellular signal-regulated kinase
- FGF2:
-
Fibroblast growth factor-2
- HFD:
-
High-fat diet
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein-1
- MEK:
-
MAPK/ERK
- oxLDL:
-
Oxidised LDL
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
PDGF receptor
- PTP:
-
Protein tyrosine phosphatase
- qPCR:
-
Quantitative real-time PCR
- RD:
-
Regular diet
- α-SMA:
-
α-Smooth muscle actin
- VSMC:
-
Vascular smooth muscle cell
References
Gallo R, Padurean A, Jayaraman T et al (1999) Inhibition of intimal thickening after balloon angioplasty in porcine coronary arteries by targeting regulators of the cell cycle. Circulation 99:2164–2170
Sobel BE (2001) Acceleration of restenosis by diabetes: pathogenetic implications. Circulation 103:1185–1187
Moses JW, Leon MB, Popma JJ et al (2003) Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med 349:1315–1323
Bonaa KH, Mannsverk J, Wiseth R et al (2016) Drug-eluting or bare-metal stents for coronary artery disease. N Engl J Med 375:1242–1252
Silber S, Serruys PW, Leon MB et al (2013) Clinical outcome of patients with and without diabetes mellitus after percutaneous coronary intervention with the resolute zotarolimus-eluting stent: 2-year results from the prospectively pooled analysis of the international global RESOLUTE program. JACC Cardiovasc Interv 6:357–368
Lamharzi N, Renard CB, Kramer F et al (2004) Hyperlipidemia in concert with hyperglycemia stimulates the proliferation of macrophages in atherosclerotic lesions: potential role of glucose-oxidized LDL. Diabetes 53:3217–3225
Raines EW, Koyama H, Carragher NO (2000) The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann N Y Acad Sci 902:39–51, Discussion 51–52
Lo HM, Wu MW, Pan SL, Peng CY, Wu PH, Wu WB (2012) Chrysin restores PDGF-induced inhibition on protein tyrosine phosphatase and reduces PDGF signaling in cultured VSMCs. J Nutr Biochem 23:667–678
Kappert K, Paulsson J, Sparwel J et al (2007) Dynamic changes in the expression of DEP-1 and other PDGF receptor-antagonizing PTPs during onset and termination of neointima formation. FASEB J 21:523–534
King GL, Wakasaki H (1999) Theoretical mechanisms by which hyperglycemia and insulin resistance could cause cardiovascular diseases in diabetes. Diabetes Care 22(suppl 3):C31–C37
Croce K, Gao H, Wang Y et al (2009) Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 120:427–436
Koyama M, Oka T, Ouchida M et al (2003) Activated proliferation of B cell lymphomas/leukemias with the SHP1 gene silencing by aberrant CpG methylation. Lab Invest 83:1849–1858
Banskota NK, Taub R, Zellner K, King GL (1989) Insulin, insulin-like growth factor I and platelet-derived growth factor interact additively in the induction of the protooncogene c-myc and cellular proliferation in cultured bovine aortic smooth muscle cells. Mol Endocrinol 3:1183–1190
Poling J, Szibor M, Schimanski S et al (2011) Induction of smooth muscle cell migration during arteriogenesis is mediated by Rap2. Arterioscler Thromb Vasc Biol 31:2297–2305
Stout RW, Bierman EL, Ross R (1975) Effect of insulin on the proliferation of cultured primate arterial smooth muscle cells. Circ Res 36:319–327
Kashishian A, Kazlauskas A, Cooper JA (1992) Phosphorylation sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. EMBO J 11:1373–1382
Danenberg HD, Welt FG, Walker M 3rd, Seifert P, Toegel GS, Edelman ER (2002) Systemic inflammation induced by lipopolysaccharide increases neointimal formation after balloon and stent injury in rabbits. Circulation 105:2917–2922
Kim SH, Ahn KS, Jeong SJ et al (2011) Janus activated kinase 2/signal transducer and activator of transcription 3 pathway mediates icariside II-induced apoptosis in U266 multiple myeloma cells. Eur J Pharmacol 654:10–16
Cheng T, Zhang Y, Zhang T, Lu L, Ding Y, Zhao Y (2015) Comparative pharmacokinetics study of Icariin and Icariside II in rats. Molecules 20:21274–21286
Geraldes P, Hiraoka-Yamamoto J, Matsumoto M et al (2009) Activation of PKC-δ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med 15:1298–1306
Jiang ZY, Lin YW, Clemont A et al (1999) Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest 104:447–457
Mima A, Kitada M, Geraldes P et al (2012) Glomerular VEGF resistance induced by PKCdelta/SHP-1 activation and contribution to diabetic nephropathy. FASEB J 26:2963–2974
Xu E, Charbonneau A, Rolland Y et al (2012) Hepatocyte-specific Ptpn6 deletion protects from obesity-linked hepatic insulin resistance. Diabetes 61:1949–1958
Kazlauskas A, Ellis C, Pawson T, Cooper JA (1990) Binding of GAP to activated PDGF receptors. Science 247:1578–1581
Palen DI, Belmadani S, Lucchesi PA, Matrougui K (2005) Role of SHP-1, Kv.1.2, and cGMP in nitric oxide-induced ERK1/2 MAP kinase dephosphorylation in rat vascular smooth muscle cells. Cardiovasc Res 68:268–277
Tibaldi E, Zonta F, Bordin L et al (2014) The tyrosine phosphatase SHP-1 inhibits proliferation of activated hepatic stellate cells by impairing PDGF receptor signaling. Biochim Biophys Acta 1843:288–298
Sedding DG, Widmer-Teske R, Mueller A et al (2013) Role of the phosphatase PTEN in early vascular remodeling. PLoS One 8, e55445
Micke P, Hackbusch D, Mercan S et al (2009) Regulation of tyrosine phosphatases in the adventitia during vascular remodelling. Biochem Biophys Res Commun 382:678–684
Kappert K, Peters KG, Bohmer FD, Ostman A (2005) Tyrosine phosphatases in vessel wall signaling. Cardiovasc Res 65:587–598
Kruger J, Wellnhofer E, Meyborg H et al (2016) Inhibition of Src homology 2 domain-containing phosphatase 1 increases insulin sensitivity in high-fat diet-induced insulin-resistant mice. FEBS Open Bio 6:179–189
Friedman AD, Claypool SE, Liu R (2013) The smart targeting of nanoparticles. Curr Pharm Des 19:6315–6329
Acknowledgements
We thank A. Marette (Montreal Diabetes Research Center, Canada) for providing the Shp-1 cDNA construct, P. Geraldes (Montreal Diabetes Research Center, Canada) for technical advice and Y. Tesmenitsky (Cardiovascular Clinical Research Center, USA), I. Wu and E. Yu (Joslin Diabetes Center, USA) for their technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work is supported by JDRF Fellowship (WQ), ADA mentor fellowship (QL), K99/R00 DK090210 (CWL), NIDDK R01DK053105-10, P30DK036836 and DRC grant (P30DK036836-30) (GLK).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
WQ and QL contributed to the conception and design of the study, acquisition, analysis and interpretation of the data and drafting the article. CWL, CRM, SL, LMR, XW, YX, MK and KC participated in the acquisition of data and critical revision of the manuscript. GLK contributed to the conception and design of the studies and analysis and interpretation of the data and wrote the manuscript. All the authors approved the version to be published. GLK is the guarantor of this work.
Additional information
Weier Qi and Qian Li contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 1107 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Qi, W., Li, Q., Liew, C.W. et al. SHP-1 activation inhibits vascular smooth muscle cell proliferation and intimal hyperplasia in a rodent model of insulin resistance and diabetes. Diabetologia 60, 585–596 (2017). https://doi.org/10.1007/s00125-016-4159-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4159-1