
Abstract
Aims/hypothesis
The nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) is an important gene regulator in glucose and lipid metabolism. Unfortunately, PPARγ-activating drugs of the thiazolidinedione class provoke adverse side effects. As recently shown, amorfrutin A1 is a natural glucose-lowering compound that selectively modulates PPARγ. In this study we aimed to characterise, in vitro, a large spectrum of the amorfrutins and similar molecules, which we isolated from various plants. We further studied in vivo the glucose-lowering effects of the so far undescribed amorfrutin B, which featured the most striking PPARγ-binding and pharmacological properties of this family of plant metabolites.
Methods
Amorfrutins were investigated in vitro by binding and cofactor recruitment assays and by transcriptional activation assays in primary human adipocytes and murine preosteoblasts, as well as in vivo using insulin-resistant high-fat-diet-fed C57BL/6 mice treated for 27 days with 100 mg kg−1 day−1 amorfrutin B.
Results
Amorfrutin B showed low nanomolar binding affinity to PPARγ, and micromolar binding to the isotypes PPARα and PPARβ/δ. Amorfrutin B selectively modulated PPARγ activity at low nanomolar concentrations. In insulin-resistant mice, amorfrutin B considerably improved insulin sensitivity, glucose tolerance and blood lipid variables after several days of treatment. Amorfrutin B treatment did not induce weight gain and furthermore showed liver-protecting properties. Additionally, amorfrutins had no adverse effects on osteoblastogenesis and fluid retention.
Conclusions/interpretation
The application of plant-derived amorfrutins or synthetic analogues thereof constitutes a promising approach to prevent or treat complex metabolic diseases such as insulin resistance or type 2 diabetes.
Similar content being viewed by others

Introduction
Faced by an alarming global increase in the incidence of obesity and type 2 diabetes, there is an urgent need to develop effective treatment and prevention strategies [1]. Besides lifestyle interventions, the application of safe preventive drugs or tailored food supplements may help to combat the current epidemic of increasing insulin resistance, a hallmark of type 2 diabetes [2].
The peroxisome proliferator-activated receptor gamma (PPARγ) plays a central role in glucose and lipid homeostasis, and pharmacological modulation of this nuclear receptor is an established strategy to treat insulin resistance and dyslipidaemia [3]. However, strongly activating PPARγ drugs such as the thiazolidinediones (TZDs) were withdrawn from the market or restricted in their prescription because they provoke adverse side effects such as weight gain, oedema, osteoporosis, cancer and heart failure [4, 5]. Rosiglitazone (RGZ; Avandia) effectively improves insulin sensitivity by fully activating PPARγ-regulated gene expression in various tissues, but this non-specific transcriptional activation appears to be linked to the unwanted side effects. However, partial agonism of PPARs by selective PPARγ modulators (SPPARγMs) can promote a differential cofactor recruitment profile, leading to specific patterns of gene expression that maintain the glucose-lowering potential of full PPARγ agonists but without inducing the side effects described above. Thus, the use of SPPARγMs is a promising approach for developing glucose-lowering agents with an acceptable safety profile [6–8]. Combined activation of PPARα, PPARβ/δ and PPARγ by a single compound has further been proposed as an innovative therapeutic approach that may result in glucose-lowering effects and improved lipid profiles with fewer side effects [9].
We recently reported the characterisation of four amorfrutins, a family of phenyl terpenoid natural products, derived from a compound library [10]. Here, we explore a wide spectrum of naturally available amorfrutins and related compounds, isolated from various natural resources. We identified amorfrutin B as a novel natural PPARγ modulator with greatly increased binding affinity compared with values recently published for other amorfrutins. Amorfrutin B shows potent glucose-lowering effects in high-fat-diet (HFD)-fed mice comparable with those of RGZ, but without inducing any of the side effects typically associated with the TZDs or other strong synthetic PPARγ activators.
Methods
Materials
Compounds were purchased as described in the electronic supplementary material (ESM) Methods.
PPAR binding, cofactor recruitment and transcriptional activation assays
Binding of natural products was quantified as described in the ESM Methods.
Cell culture
For details, see the ESM Methods.
PPARγ knockdown
Specificity of PPARγ modulation was investigated in small interfering (si)RNA-mediated PPARγ-knockdown in human primary adipocytes, as described in the ESM Methods.
RNA purification, cDNA synthesis and quantitative real-time PCR
See the ESM Methods for further details. Primer sequences are summarised in ESM Table 1.
Pharmacokinetics
Pharmacokinetic experiments were performed at Charles River (Edinburgh, UK). Briefly, 18 male C57BL/6 mice were orally given amorfrutin B, 100 mg/kg body weight, in 1% (wt/vol.) carboxymethylcellulose. Terminal blood samples were collected after 0.5, 1, 2, 4, 8 and 24 h after dosing and analysed using liquid chromatography (LC)-MS/MS.
Animal studies
Animal studies were approved by the State Office of Health and Social Affairs, Berlin, Germany, and were carried out according to internationally approved guidelines. All animals were singly housed under temperature-, humidity- and light-controlled conditions (22°C, 50% humidity, 12 h light/12 h dark cycle). Mice had ad libitum access to food and water. Mice and food were weighed regularly. Male 6 week-old C57BL/6 mice were fed for 12 weeks with an HFD (D12492, kJ composition 60% fat, 19% protein, 21% carbohydrate, 25.3 MJ/kg; ssniff, Soest, Germany) to induce obesity and insulin resistance. Prior to treatment, mice were weighed and distributed uniformly to three groups (n = 10–13 each). Mice were fed over 4 weeks with an HFD without compound (vehicle), with 4 mg kg−1 day−1 RGZ or with 100 mg kg−1 day−1 amorfrutin B incorporated in the food.
After 15 days of treatment an intraperitoneal insulin sensitivity test (IPIST) was performed. Mice were fasted overnight and then had ad libitum access to food for 1 h before the test. Insulin (Sigma-Aldrich, Taufkirchen, Germany) at 1.5 U/kg body weight was injected i.p. Blood was taken from the tail vein at the indicated time points and blood glucose was analysed in a Hemocue B-Glucose analyser (Hemocue, Großostheim, Germany). Whole blood was further collected using Microvette lithium-heparin coated capillary tubes (CB300, Sarstedt, Nürnbrecht, Germany). After centrifugation for 5 min at 2,000 g and 4°C, plasma was collected and stored at −80°C for subsequent measurements.
After 22 days of feeding an OGTT was carried out. Mice were fasted overnight before being subjected to an oral glucose dose of 2 g/kg body weight (Sigma-Aldrich). Blood was taken from the tail vein at the indicated time points and examined as described above.
After 27 days of dosing, fasted mice were killed by cervical dislocation. Hematocrit was measured by weighting of blood samples before and after plasma separation and is given in wt/wt %. Plasma and tissues were collected and stored at −80°C before use.
Metabolic variable measurements
Blood glucose was analysed in a Hemocue B-Glucose analyser. Plasma glucose was measured using the Amplex Red Glucose Assay Kit (Life Technologies, Carlsbad, CA, USA). Plasma triacylglycerols, NEFA, HDL- and LDL-cholesterol and plasma alanine transaminase (ALT) were determined with colorimetric quantification kits (Biovision, Milpitas, CA, USA). Plasma β-hydroxybutyrate was analysed colorimetrically (Cayman Chemical, Ann Arbor, MI, USA). Insulin (Insulin Ultrasensitive EIA, ALPCO, Salem, NH, USA), T3, T4 (Calbiotech, San Diego, CA, USA), osteocalcin (bone γ-carboxyglutamate protein [BGLAP], ABIN415574, antibodies-online, Aachen, Germany) and FGF21 (BioVendor, Heidelberg, Germany) were determined in plasma samples using ELISA. Whole-blood haemoglobin (Hemoglobin Colorimetric Assay Kit, Cayman Chemical) was analysed for investigation of haemodilution. All assays were performed according to the manufacturer’s instructions. HOMA-insulin resistance (IR) was determined according to HOMA-IR = fasting blood glucose (mmol/l) × fasting insulin (pmol/l)/156.
Immunoblotting
PPARγ-Ser273 phosphorylation was investigated as described in ESM Methods.
Statistical analyses
Data are presented as mean ± SEM if not otherwise denoted. Statistical tests were performed in GraphPad Prism 5.0 (La Jolla, CA, USA). For comparison of two groups, statistical significance was examined by unpaired two-tailed Student’s t test if not otherwise stated. For multiple comparisons, data were analysed by one-way ANOVA with subsequent Dunnett’s post hoc test. A p value ≤ 0.05 was defined as statistically significant. Further details are described in ESM Methods.
Results
Amorfrutin B is the most selective and potent SPPARγM of various natural amorfrutins and partially recruits important transcriptional cofactors
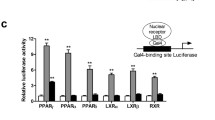
We first analysed a series of amorfrutins isolated from the edible fruits of Amorpha fruticosa and edible roots of Glycyrrhiza foetida, or other natural resources, using in vitro binding assays. In addition to the four recently described amorfrutins [10], we discovered 34 amorfrutins and related natural products from 38 candidate compounds (ESM Fig. 1) with binding affinities for PPARγ ranging from 19 nmol/l to 6 μmol/l (ESM Fig. 2 and ESM Table 2). In agreement with our structural analyses of these natural products [11], amorfrutin B (Fig. 1a) had the lowest, nanomolar, binding affinity constant (K i) to PPARγ (K i = 19 nmol/l; Table 1) similar to the standard PPARγ-targeting drug RGZ (K i = 7 nmol/l; Table 1), and 12 times lower than the initially described amorfrutin A1 [10]. Additionally, the amorfrutins bound to the subtypes PPARα and PPARβ/δ (ESM Figs 3, 4 and ESM Table 2). Amorfrutin B revealed potent binding to PPARα (K i = 2,624 nmol/l, Table 1) and PPARβ/δ (K i = 1,782 nmol/l, Table 1) with low-micromolar affinity, suggesting that this compound may also modulate the activity of these PPARs. Such partial pan-PPAR modulation may facilitate the concomitant treatment of diabetes-associated disorders, but is difficult to trace mechanistically because of the involvement, and potential cross-talk, of various tissues.

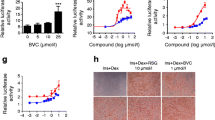
Amorfrutin B induces partial activation of PPARs. (a) Chemical structure of amorfrutin B. (b) Transcriptional activation of PPARγ by amorfrutin B (black triangles) or rosiglitazone (grey squares) in a reporter gene assay. (c) Transcriptional activation of PPARα by amorfrutin B (black triangles) or GW7647 (grey circles) in a reporter gene assay. (d) Transcriptional activation of PPARβ/δ by amorfrutin B (black triangles) or GW0742 (grey diamonds) in a reporter gene assay. (e–i) Recruitment of transcriptional cofactor peptides to PPARγ-LBD titrated with amorfrutin B (black triangles) or rosiglitazone (grey squares). Binding of cofactor peptides was measured by time-resolved fluorescence resonance energy transfer. Recruitment is represented relative to RGZ. Peptides are derived from coactivators CBP (e), PGC1α (f), TRAP220/DRIP (g), PRIP/RAP250 (h) or corepressor NCoR (i). Data are expressed as mean ± SD (n = 3)
We further explored the transcriptional activation potential of the amorfrutins using reporter gene assays. We observed PPARγ activation, with half-maximal effective concentration (EC50) values ranging from 25 nmol/l to 6.9 μmol/l and transactivation efficacies ranging from 5% to 79% relative to RGZ (ESM Fig. 5 and ESM Table 2). Once more, amorfrutin B showed nanomolar effective concentrations (EC50 = 73 nmol/l) and reduced maximal PPARγ activation (efficacy = 25%, Fig. 1b and Table 1). In contrast to the full PPARγ agonist RGZ, amorfrutins only partially induced transcriptional activation of PPARγ, suggesting that these natural products represent a new chemical class of selective PPAR modulators (SPPARM).
Transcriptional activation of the PPARα subtype was additionally tested in reporter gene assays, with amorfrutin B revealing EC50 and efficacy values of 906 nmol/l and 61%, respectively (Fig. 1c and Table 1), and a range from 157 nmol/l to 4.7 μmol/l and 13% to 68%, respectively, for the related compounds (ESM Fig. 6 and ESM Table 2). Despite activation of the PPARβ/δ subtype by amorfrutin B at nanomolar concentrations (EC50 = 740 nmol/l), efficacy of PPARβ/δ activation was low (3%; Fig. 1d and Table 1).
Furthermore, amorfrutin B only partially induced recruitment of important transcriptional cofactors including CREB-binding protein (CBP), PPARγ coactivator 1 (PGC1)α, thyroid hormone receptor-associated protein complex 220 kDa component (TRAP220)/vitamin D receptor interacting protein (DRIP) and PPAR-interacting protein (PRIP)/nuclear receptor coactivator 250 (RAP250) to PPARγ (Fig. 1e–i, Table 2 and ESM Fig. 7). In contrast, amorfrutin B reduced binding of the corepressor nuclear receptor corepressor 1 (NCoR) with an IC50 value similar to RGZ (60 nmol/l vs 23 nmol/l with RGZ), but with lower maximal dissociation efficacy (61% vs RGZ, Fig. 1i, Table 2). This was similar for other amorfrutin variants (ESM Fig. 7 and ESM Table 2). These results indicate that amorfrutin B is a high-affinity SPPARM with potential to exhibit strong glucose-lowering properties without provoking side effects associated with full PPARγ activation.
Amorfrutin B selectively activates PPARγ in primary human adipocytes
Consistent with partial PPARγ activation, in human primary adipocytes amorfrutin B induced the expression of adipogenesis-related genes such as CCAAT/enhancer binding protein α and β (CEBPA and CEBPB) and the fatty acid binding protein 4 (FABP4) much less strongly than RGZ (Fig. 2a), indicating alleviated adipocyte differentiation. In contrast to RGZ, amorfrutin B further showed reduced RNA expression of the cortisol generating hydroxysteroid (11-β) dehydrogenase 1 (HSD11B1), which is linked to central obesity [12]. Additionally, amorfrutin B treatment led to decreased transcription of pyruvate dehydrogenase kinase 4 (PDK4), encoding a glycerogenesis-activating enzyme linked to excess lipid storage in adipocytes [13]. However, amorfrutin B treatment also resulted in increased phosphodiesterase 3B (PDE3B) expression, which is responsible for beneficial NEFA release in TZD-treated mice [14]. No treatment had any effects on NCOR1 gene expression (ESM Fig. 8).

Gene expression profile of amorfrutin B and RGZ in primary human adipocytes. Cells were treated for 24 h with 10 μmol/l RGZ (grey bars) or 10 μmol/l amorfrutin B (black bars) and expression of known PPARγ target genes (a) and adipokines (b) was analysed by quantitative PCR. (c) Primary human adipocytes were transfected with PPARγ siRNA (hatched bars) or control siRNA (unhatched bars) and were treated with either 10 μmol/l RGZ (grey bars), 10 μmol/l amorfrutin B (black bars) or vehicle only (white bars) for 24 h. RNA expression was analysed by quantitative PCR. Data are expressed as mean ± SEM (n = 3–4/group). * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001 vs vehicle. AB, amorfrutin B; neg., negative; NS, not significant
As secretion of endocrine factors by adipose tissue plays a pivotal role in systemic metabolism, we further investigated the expression of important adipokines in treated human adipocytes. Amorfrutin B treatment led to increasing transcription of the beneficial adipokines adiponectin (ADIPOQ), fibroblast growth factor 21 (FGF21) and angiopoietin-like 4 (ANGPTL4), similar to RGZ treatment, but we observed no regulation of leptin (LEP) transcription (Fig. 2b). Furthermore, amorfrutin B resulted in reduced expression of pro-inflammatory angiopoietin-like 2 (ANGPTL2), retinoic acid receptor responder 2 (RARRES2) and adipsin (complement factor 2 [CFD]) (Fig. 2b). In summary, these data indicate potential glucose-lowering effects with, presumably, lower susceptibility to adipose-derived body weight gain in vivo with amorfrutin B treatment.
Knockdown of PPARγ in these cells led to a significant reduction of amorfrutin B-induced gene expression (Fig. 2c), corroborating specific modulation of PPARγ-derived transcription in human adipocytes.
Pharmacokinetic profile of amorfrutin B in C57BL/6 mice
To start exploring the pharmacological profile of amorfrutin B, C57BL/6 mice were orally challenged once with a loading dose of 100 mg/kg amorfrutin B (Fig. 3a, Table 3). Administration of amorfrutin B led to a fast plasma concentration peak, indicating a high bioavailability of that natural product. Amorfrutin B was almost completely eliminated within 24 h of the dose, with an elimination half-life of about 2 h (Table 3). Amorfrutin B and amorfrutin A1 [10] showed similar pharmacokinetic properties (Table 3).

Effect of amorfrutin B treatment in insulin-resistant DIO mice. (a) Pharmacokinetic profile of amorfrutin B after oral administration in C57BL/6 mice. Data are expressed as mean ± SEM (n = 3). (b) Fasting blood glucose of DIO mice after 15 days of treatment with vehicle (body weight = 41.7 ± 1.1 g), RGZ (body weight = 41.2 ± 1.5 g) or amorfrutin B (body weight = 35.7 ± 1.4 g). (c) Fasting plasma insulin of DIO mice after 15 days of treatment. (d) Effect of treatment for 15 days on insulin resistance determined by HOMA-IR. (e, f) Glucose and insulin concentrations during OGTT after 22 days of treatment with vehicle (white circles/bars, body weight = 39.9 ± 1.0 g [n = 13]), RGZ (grey squares/bars, body weight = 39.9 ± 1.5 g [n = 8–10]) or amorfrutin B (black triangles/bars, body weight = 34.4 ± 1.3 g [n = 13]). (g) Glucose levels during the IPIST after 15 days of treatment. Data are expressed as mean ± SEM. * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001 vs vehicle. AB, amorfrutin B; AUCi, inverse AUC; VEH, vehicle
Amorfrutin B has strong glucose-lowering effects and considerably improves dyslipidaemia in mice with diet-induced obesity
To characterise amorfrutin B in a diabetic obese animal model, we treated HFD-fed C57BL/6 mice for 4 weeks with 100 mg kg−1 day−1 amorfrutin B, 4 mg kg−1 day−1 RGZ or vehicle only. After 2 weeks amorfrutin B-treated mice exhibited reduced concentrations of fasting blood glucose (Fig. 3b) and fasting plasma insulin (Fig. 3c), equal to or more reduced than in RGZ-treated mice. Amorfrutin B- and RGZ-treated mice showed equal reductions in insulin resistance as determined by homeostatic modelling (Fig. 3d). In OGTT (Fig. 3e, f) and IPIST (Fig. 3g) amorfrutin B treatment resulted in reductions in insulin resistance and glucose intolerance similar to those achieved with RGZ.
Amorfrutin B strongly decreased the concentration of triacylglycerols and NEFA in the plasma of HFD-fed mice, comparable with RGZ (Fig. 4). After 4 weeks of treatment, amorfrutin B and RGZ both reduced fasting plasma levels of triacylglycerols by 25% (Fig. 4a). Fasting concentrations of deleterious plasma NEFA were decreased by 29% with amorfrutin B and 40% with RGZ treatment (Fig. 4b).

Effect of amorfrutin B treatment on plasma lipid variables in insulin-resistant DIO mice. (a) Fasting plasma triacylglycerols after 27 days of treatment. (b) Fasting plasma NEFA after 27 days of treatment. Data are expressed as mean ± SEM. * p ≤ 0.05 and ** p ≤ 0.01 vs vehicle. Vehicle, n = 13; RGZ, n = 10; amorfrutin B, n = 12–13. AB, amorfrutin B; VEH, vehicle
Based on the results described above, the physiological effects of amorfrutin B treatment seemed to be largely controlled via nanomolar activation of PPARγ and differential expression of its target genes in the adipocytes. In visceral white adipose tissue (WAT), amorfrutin B significantly increased the expression of PGC1α and PGC1β (Ppargc1a and Ppargc1b) (twofold each, ESM Fig. 9), indicating improved mitochondrial biogenesis and fatty acid breakdown. Amorfrutin B and RGZ equally increased expression of adiponectin (Adipoq) and uncoupling protein 3 (Ucp3), by approximately twofold each, but only RGZ clearly boosted Ucp1 expression (20-fold vs 1.6-fold, ESM Fig. 9).
We investigated further in vivo gene expression to explore potential additional physiological effects derived from regulation via the liver and skeletal muscle. In liver, in contrast to RGZ, amorfrutin B only partly regulated gluconeogenic genes (ESM Fig. 10a). Both RGZ and amorfrutin B showed a 25% increase in transcription of Ppargc1a but in general amorfrutin B treatment resulted in minor changes in the expression of fatty acid oxidation genes (ESM Fig. 10b). The most prominent difference between RGZ- and amorfrutin B-treated livers was pyruvate dehydrogenase 4 (Pdk4), which was highly activated only in RGZ-treated mice (+96% vs vehicle, ESM Fig. 10b). These data confirm the effects observed in human primary adipocytes (Fig. 2a), suggesting a switch between carbohydrate and lipid metabolism that may play a role in the different mechanisms between RGZ and amorfrutin B.
As ketone body metabolism is involved in diabetes and obesity, we further analysed the expression of ketogenic genes in the livers of these mice. Both RGZ and amorfrutin B activated genes of ketone metabolism, with a striking increase in expression of 3-hydroxy-3-methylglutaryl-CoA synthase 1 (Hmgcs1) in amorfrutin B-treated mice (+123%, ESM Fig. 10c). However, plasma levels of β-hydroxybutyrate were not significantly changed after treatment (ESM Fig. 10d). To further explore potential PPARβ/δ-induced insulin-sensitising effects in skeletal muscle, we analysed the expression of various target genes but could not detect any amorfrutin-induced differential regulation (ESM Fig. 10e), suggesting a minor role of the skeletal muscle in the observed glucose-lowering profile of amorfrutins, which is in agreement with the low PPARβ/δ activation observed in reporter gene assays (Fig. 1d).
To investigate the role of the peptide hormone fibroblast growth factor 21 (FGF21) in the observed metabolic improvement, we additionally determined Fgf21 expression in WAT and liver, and circulating FGF21 plasma levels. As described for TZDs [15], RGZ induced Fgf21 gene expression in liver and adipose tissue (ESM Fig. 11) and strikingly increased FGF21 plasma levels (ESM Fig. 11). However, amorfrutin B completely failed to induce expression and secretion of FGF21, suggesting alternative PPARγ activation to the described TZD–FGF21 autocrine loop. We further explored if the phosphorylation of PPARγ on Ser273 in WAT [16] might mediate the glucose-lowering effects of amorfrutin B, but did not observe any significant change at this phosphorylation site after RGZ or amorfrutin B treatment (ESM Fig. 12).
Amorfrutin B does not induce the adverse side effects associated with TZDs
As discussed above, PPARγ-targeting drugs such as RGZ are linked to undesirable side effects. We therefore tested for potential side effects of amorfrutin B treatment. Viability and cytotoxicity assays in HepG2 cells did not reveal any adverse effects up to 32 μmol/l amorfrutin B (ESM Fig. 13). The potential carcinogenicity of amorfrutin B was evaluated using a cellular micronucleus assay. In this assay, the formation of micronuclei during cell division of Chinese hamster ovary (CHO) cells is observed microscopically after treatment with potential mutagens. Amorfrutin B was tested up to a concentration of 32 μmol/l and showed no genetic toxicity either in the presence (+S9) or absence (−S9) of metabolic activation by extracts of rat liver homogenate (Fig. 5a). Of note, amorfrutin B significantly reduced the basal formation of micronuclei in the presence of S9 extract in a concentration-dependent manner, thus improving genetic integrity. After 4 weeks of treatment, the livers of RGZ-treated HFD-fed obese mice showed strongly increased expression of inflammatory markers, indicating macrophage infiltration and potential local inflammation. In contrast, amorfrutin B treatment led to reduced gene expression of these markers (Fig. 5b), suggesting anti-inflammatory effects of amorfrutin B treatment in the livers of HFD-fed mice. In addition, RGZ treatment increased Fabp4 expression by a factor of 31, indicating increased lipid storage in the mouse liver, whereas amorfrutin B did not show any increase in Fabp4 expression (Fig. 5c). Consistently, RGZ elevated plasma ALT levels compared with untreated HFD-fed mice, indicating liver toxicity. In contrast, amorfrutin B treatment led to a reduction of plasma ALT levels (Fig. 5d), in agreement with the liver-protective effects previously reported for amorfrutin A1 [10].

Amorfrutins do not provoke side effects associated with classic PPARγ agonists. (a) Analysis of genetic toxicity in the in vitro micronucleus assay in CHO cells in the absence (−S9) and presence (+S9) of rat liver homogenate extract. CHO cells were treated with different concentrations of amorfrutin B (n = 2), the clastogens mitomycin C (n = 4) or cyclophosphamide (n = 4) or vehicle, and micronuclei were stained with Hoechst dye and counted by fluorescence microscopy. (b) The livers of DIO mice treated for 4 weeks with RGZ (grey) or amorfrutin B (black) were analysed by quantitative PCR for expression of genes associated with macrophage invasion and inflammation (n = 6–9 per group). (c) RNA expression of liver Fabp4 was analysed by quantitative PCR. (d) Effect of amorfrutin B (n = 13) and RGZ (n = 10) or vehicle (n = 11) on liver-damage-indicating plasma levels of ALT after treatment for 4 weeks in DIO mice. (e) MC3T3-E1 preosteoblasts (hatched bars) were differentiated to osteoblasts in the presence of vehicle only (white, set to 1), RGZ (grey) or amorfrutin B (black). The expression of genes involved in osteoblastogenesis was determined by quantitative PCR. (f) Calcification of differentiated MC3T3-E1 osteoblasts treated with amorfrutin B (n = 6), RGZ (n = 4) and vehicle (n = 4) was measured by Alizarin Red S staining. (g) The effect of amorfrutin B on plasma osteocalcin concentration after treatment for 4 weeks in DIO mice. Osteocalcin was measured by ELISA. (h) Body weight gain in obese mice after treatment with amorfrutin B (black triangles, n = 13), RGZ (grey squares, n = 10) and vehicle only (white circles, n = 13). Data are expressed as mean ± SEM. * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001 vs vehicle. AB, amorfrutin B; CP, cyclophosphamide; MMC, mitomycin C; NS, not significant; PRE, preosteoblasts; VEH, vehicle
Another well-known side effect of TZDs is the impairment of osteoblastogenesis, leading to osteoporosis and increased fracture risk. Treatment of murine MC3T3-E1 preosteoblasts with RGZ or related TZDs resulted in reduced expression of genes involved in osteoblastogenesis, such as phosphate-regulating gene with homologies to endopeptidases on the X chromosome (Phex), integrin-binding sialoprotein (Ibsp) and osteocalcin (Bglap) (Fig. 5e). RGZ treatment led to impaired calcification of bone cells in vitro as assessed by Alizarin Red S staining (Fig. 5f). In contrast, treatment with amorfrutin B and related amorfrutins did not show any reductions in expression of the osteoblast gene set (Fig. 5e and ESM Fig. 14a) or in mineralisation of bone cells (Fig. 5f and ESM Fig. 14b) in vitro. In HFD-fed mice, RGZ increased plasma bone osteocalcin levels by 50% (Fig. 5g), indicating elevated bone cell turnover. However, there was no effect on plasma bone osteocalcin levels in amorfrutin B-treated diet-induced obesity (DIO) mice (Fig. 5g). Taken together, these results indicate that amorfrutins do not provoke osteoporosis in mice.
Adverse body weight gain is a frequent side effect of PPARγ activation due to increased fat storage in WAT. However, in HFD-fed mice amorfrutin B treatment did not lead to increased adiposity but, instead, to a beneficial reduction of body weight gain by approximately 15% compared with HFD-fed mice treated with vehicle control (Fig. 5h). Given the inconspicuous results from the various described assays, this observation cannot be explained by potential compound toxicity. Feeding of amorfrutin B only had a slight impact on food intake during the first 3 days (ESM Fig. 15a), and correction for food intake using analysis of covariance (ANCOVA, ESM Fig. 15b) revealed that anoretic effects cannot wholly explain the observed reduction in weight gain after amorfrutin B treatment. To investigate the potential contribution of the hypothalamic–pituitary–thyroid axis we measured plasma levels of T3 and T4. Amorfrutin B-treated mice did not show significant changes in these endocrine hormones (ESM Fig. 15c). In general, weight reduction may result in improved insulin sensitivity. Although amorfrutin B-induced effects on body weight correlated (in part) with improved insulin sensitivity, ANCOVA clearly showed glucose-lowering effects that were independent from weight regulation (ESM Fig. 15d).
RGZ has recently been reported to decrease HDL-cholesterol [17]. After 4 weeks of treatment, RGZ-treated DIO mice showed a 24% reduction in plasma HDL-cholesterol, whereas amorfrutin B changed neither plasma HDL-cholesterol nor LDL-/VLDL-cholesterol levels (ESM Fig. 16a).
RGZ administration is further associated with the development of haemodilution and oedema as a result of fluid retention. Amorfrutin B treatment did not impair haematocrit (ESM Fig. 16b) or levels of whole-blood haemoglobin (ESM Fig. 16c) in DIO mice; however, in our hands RGZ-treated mice did not show any signs of fluid retention after 4 weeks of treatment.
In summary, these data suggest that natural amorfrutins are well-tolerated glucose-lowering agents without the side effects typically associated with clinically used synthetic PPARγ agonists.
Discussion
We explored the potential of a series of naturally occurring amorfrutins, including molecules that have not been described to date. To our knowledge, amorfrutin B is the plant molecule with the highest reported binding affinity for PPARγ, allowing for efficient and selective PPARγ activation. Treatment with amorfrutins resulted in beneficial glucose-lowering effects in a murine model of diet-induced insulin resistance. Compounds derived from natural resources may have additional application areas compared with synthetic molecules. Our results provide evidence that natural extracts of amorfrutins derived from edible biomaterials such as liquorice could potentially be developed as phytomedicals or functional food products for the prevention of insulin resistance.
Interestingly, amorfrutin B did not provoke any of the side effects that are commonly associated with TZDs. PPARγ agonists such as the TZDs induce weight gain in mice and humans, a side effect that is counterproductive, particularly given the causal link between insulin resistance and obesity [2, 18]. In this study, we mainly focused on the glucose-lowering effects of amorfrutin B. The observed weight-reducing effects of amorfrutin B might also have contributed to the improvement of insulin sensitivity. The correction for body weight by using ANCOVA [19] unambiguously demonstrated that weight-reducing effects alone cannot account for the observed glucose-lowering effects of amorfrutin B treatment. Correspondingly, we could show that in leptin-receptor-deficient db/db mice amorfrutin A1 markedly improved insulin sensitivity without affecting body weight [10].
In general, weight regulation can be caused by various poorly understood mechanisms, including cross-talk between many tissues. Decreased energy intake was only a minor observed effect in our study (ESM Fig. 15a–b). Elevated energy expenditure controlled via the hypothalamic–pituitary–thyroid axis can be a main contributor to overweight loss. However, using amorfrutin B, neither T3 nor T4 plasma levels were significantly increased in DIO mice (ESM Fig. 15c). Energy expenditure can be enhanced by increased physical activity and/or thermogenesis. Amorfrutin-induced activation of PPARβ/δ, albeit low, may have resulted in thermogenic effects. For example, in the WAT of amorfrutin B-treated DIO mice we observed a significant activation of expression of Ppargc1a, Ppargc1b and Ucp3, and a marginal increase in Ucp1 expression (ESM Fig. 9), which might have contributed to increased energy expenditure. However, these effects were not unique to amorfrutin-treated mice, as RGZ induced a similar profile with the exception of an additional striking boost in Ucp1 transcription. Future experiments will focus on the dissection of multifactorial weight-reducing effects of amorfrutins.
The peptide hormone FGF21 was shown to integrate several aspects of metabolism in the pathophysiology and pharmacology of metabolic disorders [20, 21], suggesting a mechanistic link between the effects of administered PPARγ TZD ligands and FGF21. As Fgf21-knockout mice were refractory to both the beneficial insulin-sensitising effects and the detrimental side effects of RGZ, FGF21 might be a key mediator of PPARγ actions. However, in amorfrutin B-treated mice, systemic FGF21 plasma levels as well as local Fgf21 gene expression were unchanged (ESM Fig. 11). The fact that amorfrutin B seems to activate PPARγ and improve insulin sensitivity without significant interaction with FGF21 or without inhibiting the phosphorylation of PPARγ-Ser273 necessitates further mechanistic studies and validation, including the search for additional post-translational modifications of PPARγ and its cofactors.
In addition to the glucose-lowering effects, amorfrutin B considerably lowered plasma levels of ALT, indicating liver-protecting properties of amorfrutins. As we have shown recently, the amorfrutin variant A1 prevented the formation of liver steatosis in HFD-fed C57BL/6 mice through induction of fatty acid oxidation and anti-inflammatory gene expression profiles [10]. These diverse effects might have occurred due to simultaneous, more or less efficient activation of PPARγ, PPARα and PPARβ/δ (Table 1). As a single oral dose of 100 mg/kg in DIO mice leads to a maximum plasma concentration of 30.4 mg/l and a mean concentration of 3.7 mg/l over 24 h (Fig. 3a, Table 3), activation of PPARα and PPARβ/δ is of potential physiological relevance and might contribute to systemic effects in tissues such as the liver. Consequently, amorfrutin B and related natural molecules might be considered partial pan-PPAR agonists with potentially synergistic effects.
Dietary small molecules derived from food ingredients of various cultures still represent a largely unexplored resource for preventing or treating complex diseases such as insulin resistance. The identification of the potent natural PPARγ-activator amorfrutin B, which can be isolated in large quantities from liquorice, highlights the medicinal potential of natural dietary substances.
Abbreviations
- ALT:
-
Alanine transaminase
- BGLAP:
-
Bone γ-carboxyglutamate protein
- CBP:
-
CREB-binding protein
- CHO:
-
Chinese hamster ovary
- DIO:
-
Diet-induced obesity
- DRIP:
-
Vitamin D receptor interacting protein
- EC50 :
-
Half-maximal effective concentration
- HFD:
-
High-fat diet
- HOMA-IR:
-
HOMA of insulin resistance
- IPIST:
-
Intraperitoneal insulin sensitivity test
- K i :
-
Binding affinity constant
- NCoR:
-
Nuclear receptor corepressor
- PGC1:
-
PPARγ coactivator 1
- PPAR:
-
Peroxisome proliferator-activated receptor
- PRIP:
-
PPAR-interacting protein
- RAP250:
-
Nuclear receptor coactivator RAP250
- RGZ:
-
Rosiglitazone
- Si:
-
Small interfering
- SPPAR[γ]M:
-
Selective peroxisome proliferator-activated receptor [γ] modulator
- TRAP220:
-
Thyroid hormone receptor-associated protein complex 220 kDa component
- TR-FRET:
-
Time-resolved fluorescence resonance energy transfer
- TZD:
-
Thiazolidinedione
- WAT:
-
White adipose tissue
References
Smyth S, Heron A (2006) Diabetes and obesity: the twin epidemics. Nat Med 12:75–80
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Berger J, Moller DE (2002) The mechanisms of action of PPARs. Ann Rev Med 53:409–435
(2010) Regulators restrict Avandia in the US and suspend it in the EU. Nat Rev Drug Discov 9:828–829
Rosen CJ (2010) Revisiting the rosiglitazone story—lessons learned. N Engl J Med 363:803–806
Argmann CA, Cock TA, Auwerx J (2005) Peroxisome proliferator-activated receptor gamma: the more the merrier? Eur J Clin Investig 35:82–92, discussion 80
Cock TA, Houten SM, Auwerx J (2004) Peroxisome proliferator-activated receptor-gamma: too much of a good thing causes harm. EMBO Rep 5:142–147
Gronemeyer H, Gustafsson JA, Laudet V (2004) Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3:950–964
Pourcet B, Fruchart JC, Staels B, Glineur C (2006) Selective PPAR modulators, dual and pan PPAR agonists: multimodal drugs for the treatment of type 2 diabetes and atherosclerosis. Expert Opin Emerg Drugs 11:379–401
Weidner C, de Groot JC, Prasad A et al (2012) Amorfrutins are potent antidiabetic dietary natural products. Proc Natl Acad Sci USA 109:7257–7262
de Groot JC, Weidner C, Krausze J, et al. (2013) Structural characterization of amorfrutins bound to the peroxisome proliferator-activated receptor gamma. J Med Chem. 56:1535–1543
Masuzaki H, Paterson J, Shinyama H et al (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294:2166–2170
Cadoudal T, Distel E, Durant S et al (2008) Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes 57:2272–2279
Tang Y, Osawa H, Onuma H, Nishimiya T, Ochi M, Makino H (1999) Improvement in insulin resistance and the restoration of reduced phosphodiesterase 3B gene expression by pioglitazone in adipose tissue of obese diabetic KKAy mice. Diabetes 48:1830–1835
Muise ES, Azzolina B, Kuo DW et al (2008) Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol Pharmacol 74:403–412
Choi JH, Banks AS, Estall JL et al (2010) Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466:451–456
Shetty C, Balasubramani M, Capps N, Milles J, Ramachandran S (2007) Paradoxical HDL-C reduction during rosiglitazone and fibrate treatment. Diabet Med 24:94–97
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445
Tschop MH, Speakman JR, Arch JR et al (2012) A guide to analysis of mouse energy metabolism. Nat Methods 9:57–63
Qiang L, Accili D (2012) FGF21 and the second coming of PPARgamma. Cell 148:397–398
Dutchak PA, Katafuchi T, Bookout AL et al (2012) Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell 148:556–567
Acknowledgements
We would like to thank J. C. de Groot, K. Buessow (both HZI, Braunschweig, Germany), and A. Schoop (Consulting Biotec & Pharma R&D, Neuried, Germany) for fruitful discussions and suggestions. We are deeply grateful to K. Hansen, C. Franke, U. Schroeder and L. Hartmann (all MPI for Molecular Genetics, Berlin, Germany) for technical assistance during the animal studies.
Funding
Our work is supported by the German Ministry for Education and Research (BMBF, grant number 0315082), the National Genome Research Net (NGFN, grant number 01 GS 0828), the European Union (FP7/2007–2013, under grant agreement no. 262055 [ESGI]), the TRIAD Foundation (to F. C. Schroeder) and the Max Planck Society. This work is part of the PhD thesis of C. Weidner.
Duality of interest
K. Siems and L. Müller-Kuhrt are employees of AnalytiCon Discovery, a company that sells natural products. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
CW designed and executed the experiments, analysed and interpreted data and wrote the manuscript. SJW, AW, AF and MK contributed to experimental planning, acquisition of data and drafting of the article. KK and FCS performed the chemical synthesis of amorfrutins and contributed to the study conception and writing of the article. KK and FCS performed the chemical synthesis of amorfrutins and contributed to the study conception and writing of the article. KS and LM-K isolated the natural products, contributed to the study conception and the article revision. SS designed and coordinated the study, supervised the execution of the experiments and interpretation of the data, provided means and wrote the manuscript. All authors have approved the final version of the article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 403 kb)
ESM Fig. 2
(PDF 81 kb)
ESM Fig. 3
(PDF 91 kb)
ESM Fig. 4
(PDF 91 kb)
ESM Fig. 5
(PDF 52 kb)
ESM Fig. 6
(PDF 72 kb)
ESM Fig. 7
(PDF 91 kb)
ESM Fig. 8
(PDF 7 kb)
ESM Fig. 9
(PDF 33 kb)
ESM Fig. 10
(PDF 42 kb)
ESM Fig. 11
(PDF 68 kb)
ESM Fig. 12
(PDF 61 kb)
ESM Fig. 13
(PDF 8 kb)
ESM Fig. 14
(PDF 49 kb)
ESM Fig. 15
(PDF 116 kb)
ESM Fig. 16
(PDF 69 kb)
ESM Methods
(PDF 91 kb)
ESM Table 1
(PDF 7 kb)
ESM Table 2
(PDF 32 kb)
Rights and permissions
About this article
Cite this article
Weidner, C., Wowro, S.J., Freiwald, A. et al. Amorfrutin B is an efficient natural peroxisome proliferator-activated receptor gamma (PPARγ) agonist with potent glucose-lowering properties. Diabetologia 56, 1802–1812 (2013). https://doi.org/10.1007/s00125-013-2920-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2920-2