
Abstract
Epigenetic alterations are associated with all aspects of cancer, from tumor initiation to cancer progression and metastasis. It is now well understood that both losses and gains of DNA methylation as well as altered chromatin organization contribute significantly to cancer-associated phenotypes. More recently, new sequencing technologies have allowed the identification of driver mutations in epigenetic regulators, providing a mechanistic link between the cancer epigenome and genetic alterations. Oncogenic activating mutations are now known to occur in a number of epigenetic modifiers (i.e. IDH1/2, EZH2, DNMT3A), pinpointing epigenetic pathways that are involved in tumorigenesis. Similarly, investigations into the role of inactivating mutations in chromatin modifiers (i.e. KDM6A, CREBBP/EP300, SMARCB1) implicate many of these genes as tumor suppressors. Intriguingly, a number of neoplasms are defined by a plethora of mutations in epigenetic regulators, including renal, bladder, and adenoid cystic carcinomas. Particularly striking is the discovery of frequent histone H3.3 mutations in pediatric glioma, a particularly aggressive neoplasm that has long remained poorly understood. Cancer epigenetics is a relatively new, promising frontier with much potential for improving cancer outcomes. Already, therapies such as 5-azacytidine and decitabine have proven that targeting epigenetic alterations in cancer can lead to tangible benefits. Understanding how genetic alterations give rise to the cancer epigenome will offer new possibilities for developing better prognostic and therapeutic strategies.
Similar content being viewed by others

INTRODUCTION
Cancer is an evolutionary process through which dysregulation in select cellular mechanisms confers a clonal advantage, leading to tumor growth and eventually, metastasis. In fact, nearly all cancers are defined by several “hallmark” capabilities, including resisting cell death, evading growth suppressors, uncontrolled proliferation, neoangiogenesis, invasion/metastasis, and replicative immortality (Hanahan and Weinberg, 2011). Aberrant control of other processes, such as defective differentiation and DNA damage repair, is also linked to tumor formation. Research efforts have traditionally focused on genetic abnormalities underlying malignant transformation, due to initial technological constraints and our limited understanding of other heritable patterns of gene regulation. Early studies on these genetic alterations—copy number variations, mutations, gene rearrangements—have defined mechanisms of oncogenesis, led to the creation of targeted therapies, and improved patient outcomes for certain cancers. Only recently has it become evident that many genetic alterations in cancer target epigenetic regulators, causing cancer-associated phenotypes via epigenetic dysfunction.
Since the first discovery of cancer-associated loss of DNA methylation, the field of cancer epigenetics has grown remarkably and helped elucidate aspects of cancer biology where genetic explanations alone are insufficient (Feinberg and Vogelstein, 1983). Epigenetics is the process by which cells encode non-genetic, heritable information through alterations that do not change the DNA sequence. Generally, chromatin exists in two main forms—condensed, transcriptional silent heterochromatin and euchromatin, which is transcriptionally active. The functional unit of chromatin is the nucleosome, which is an octameric structure composed of two histones each of H2A, H2B, H3, and H4 encircled by 147 bp of DNA (Margueron and Reinberg, 2010). Regulation of the chromatin state is achieved through DNA methylation, chromatin remodeling, and/or covalent histone modifications, such as methylation, acetylation, phosphorylation, ubiquitination, and sumoylation. The major effectors of these modifications are the chromatin modifier enzymes, highly specific proteins that catalyze the addition or removal of functional groups to DNA or histone tails. These modifications alter chromatin structure through noncovalent interactions within and between nucleosomes, leading to changes in macromolecular organization and promoter accessibility. In addition, these chromatin “marks” serve as signals to other specialized proteins involved in chromatin organization, gene transcription, genome maintenance, and replication (Kouzarides, 2007; Sharma et al., 2010). As a result, aberrations in one or more of these modifiers can have profound effects on normal cell physiology and are now well-documented in many diseases, including cancer (Fig. 1).

Epigenetic regulation of gene expression. Epigenetic processes include DNA methylation, covalent histone modifications (e.g. methylation, acetylation), and chromatin remodeling (SWI/SNF complex). Modifier proteins with frequent driver mutations in cancer are shown by specific function and target site. Green and red font colors represent histone “writers” and “erasers,” respectively. Lightning bolts represent cancer-associated mutations in histones H3.1 (K27) and H3.3 (K27, G34)
It has been known for decades that epigenetic dysregulation occurs in cancer. Aberrant DNA hyper- and hypo-methylation was one of the first molecular features noted to be present in cancer cells (Feinberg and Vogelstein, 1983; Gama-Sosa et al., 1983; Rubery and Newton, 1973; Timp and Feinberg, 2013). However, the nature of epigenetic alterations and the molecular underpinnings of these changes are just beginning to be appreciated. The recent accelerated search for mutations in cancers, facilitated through next generation sequencing technologies, has greatly accelerated our understanding of the mechanisms underlying epigenetic dysfunction in cancers.
Genome-wide and exomic sequencing data from recent years have demonstrated that mutations in epigenetic modifiers comprise a large portion of all genetic events in many cancers, including tumors such as renal carcinoma, adenoid cystic carcinoma (ACC), and transitional cell carcinoma (TCC) of the bladder (Dalgliesh et al., 2010; Gui et al., 2011; Ho et al., 2013). In fact, the main objective of these recent genomic analyses is the identification of bona fide “driver” mutations in cancer genes. These mutations are defined by their ability to promote or “drive” tumorigenesis and are therefore positively selected for in the development of cancer. In contrast, “passenger” mutations, which comprise the majority of mutations identified, represent genetic events that have no direct or indirect effect on the selective growth advantage of the cell in which it occurred (Stratton et al., 2009; Vogelstein et al., 2013). Candidate epigenetic driver genes have been identified as either mutated across many cancers (e.g. KDM6A in over 12 cancers), having highly recurrent mutations (e.g. IDH1R132), or having highly prevalent mutations in select tumor histologies (e.g. MLL2 in follicular lymphoma) (You and Jones, 2012). The characterization of these driver gene mutations has enhanced our understanding of the mechanisms contributing to oncogenesis, allowed for improved prognostic assessment, and opened the door for the development of exciting new therapies. In this review, we highlight recent developments, new discoveries, and therapeutic advances involving cancer-associated mutations in epigenetic regulators.
DNA METHYLATION AND HYDROXYMETHYLATION
DNA methylation plays a well-defined role in both development and disease, including cancer. First identified in 1975, CpG island (CGI) methylation was shown to function as a relatively stable alteration on DNA that can serve to silence gene transcription (Holliday and Pugh, 1975; Riggs, 1975). We now understand that DNA methylation is much more dynamic and complex, with diverse epigenetic consequences linked to varied genomic locations of where this mark occurs. For example, DNA methylation at gene promoter CGIs potently blocks the initiation of transcription, whereas methylation within CpG-poor gene bodies may actually facilitate elongation and influence patterns of alternate splicing. In addition, DNA methylation is frequently found in repeat-rich areas of the genome and is vital for both chromosomal and genomic stability, possibly through the repression of retroviral transposons (Jones, 2012). Still, the role for this epigenetic mark at other regulatory regions, such as enhancers and insulators, has yet to be determined. Regardless, aberrant methylation in human cancer is a defining feature, with global promoter CGI hypermethylation and non-CGI hypomethylation widely reported (Ehrlich, 2002; Sharma et al., 2010). Furthermore, local variations in methylation at only several key loci have been shown to be sufficient for tumorigenesis (Lee et al., 2008; Poage et al., 2011). Importantly, these altered patterns of DNA epigenetic marks (e.g. 5-mC, 5-hmC) are frequently accompanied by a critical imbalance in transcriptional programs involving differentiation and stem cell maintenance, thereby initiating tumorigenesis and sustaining growth (Jones and Baylin, 2007).
DNA methylation can function to silence tumor suppressor genes along with genetic mutations (Herman and Baylin, 2003). For example, in the case of hereditary gastric cancer, methylation of CDH1 (which encodes the E-cadherin tumor suppressor) can function as a “second hit” and cause gastric cancer when the first allele is mutated (Grady et al., 2000). In sporadic cancers, tumor suppressor genes that are mutated in hereditary versions of the disease are frequently silenced by DNA methylation instead (Esteller et al., 2001). For example, in hereditary nonpolyposis colon cancer (HNPCC), MLH1 inactivation via mutation can lead to microsatellite instability (MSI) and tumorigenesis, whereas in sporadic colon cancers, MLH1 is frequently silenced by methylation (Kane et al., 1997; Veigl et al., 1998). These data and others indicate that aberrant DNA methylation can work along with genetic alterations to promote tumorigenesis.
DNMT3A
DNA methylation is carried out by the mammalian DNA methyltransferases (DNMTs), essential enzymes that catalyze the addition of a methyl group to cytosine in CpG dinucleotides in DNA (Jones, 2012). The conversion of 5-cytosine (5-C) to methyl-cytosine (5-mC) requires the presence of a methyl donor, S-adenosylmethionine (SAM), and one of the following catalytically active DNMTs: DNMT1, DNMT3A, or DNMT3B (Shen et al., 1992). Although there is some evidence for overlapping roles, DNMT3A and DNMT3B are essential for de novo DNA methylation, whereas DNMT1 “maintains” heritable methylation patterns across the genome during cell replication (Hsieh, 1999). In fact, the role of DNMT3A in de novo methylation was initially characterized in the context of epigenetic silencing during development, including at imprinted loci in germ cells (Kaneda et al., 2004; Okano et al., 1999). The DNMTs have long been suspected to play a role in oncogenesis, as well. Aberrations in DNA methylation—both hyper- and hypo-methylation—have been well-documented in patient tumors and cell lines for decades (El-Osta, 2004). Additionally, Oka and colleagues first showed that in some cases, DNMT3A and DNMT3B, not DNMT1, mediate the cytotoxic effects of 5-aza-dC, a therapeutic mainstay in the treatment of several hematopoietic malignancies (Oka et al., 2005; Plimack et al., 2007). Not long after, DNMT3A and DNMT3B were further implicated in both hematopoietic stem cell (HSC) renewal and differentiation, two tightly regulated processes whose perturbation can lead to carcinogenesis (Challen et al., 2012; Tadokoro et al., 2007).
DNMT3A somatic mutations were first discovered by Yamashita and colleagues following sequencing of tissue from adult patients with de novo acute myeloid leukemia (AML) (Yamashita et al., 2010). Soon after, DNMT3A mutations were reported in AML cohorts from others, with frequencies as high as 22.1% (Ley et al., 2010). The majority of mutations occur at R882 (60%–64%), and almost all are heterozygous (Ley et al., 2010; Thol et al., 2011a). DNMT3A mutations are enriched in AML patients with intermediate-risk cytogenetics and normal karyotype (Lin et al., 2011; Patel et al., 2012). They are also associated with increased age, M4 and M5 AML subtypes, worse overall survival (OS) and relapse-free survival (RFS), and increased blasts at diagnosis (Hou et al., 2012; Ley et al., 2010; Lin et al., 2011; Marcucci et al., 2012; Thol et al., 2011a; Yan et al., 2011). There is also evidence that DNTM3A mutations may be a prognostic marker for decreased time to treatment failure (TTF), duration of complete remission (CR), and disease-free survival (DFS), at least in cytogenetically normal AML (Marcucci et al., 2012; Thol et al., 2011a; Yan et al., 2011). These studies have also identified several frequently co-occurring mutations, which include NPM1, FLT3, IDH1/2, and less commonly, TET2 (Hou et al., 2012; Ley et al., 2010; Lin et al., 2011; Marcucci et al., 2012; Renneville et al., 2012; Thol et al., 2011a; Yan et al., 2011). As alterations in epigenetic regulators frequently lead to genomic instability, DNMT3A mutation may further drive progression of the disease itself (Wakita et al., 2013). Further, recent experimental work has demonstrated that DNMT3A mutations at R882 are only found in major clones, suggesting this genetic alteration may be an initiating lesion in AML (Bisling et al., 2013).
DNMT3A mutations have also been described, albeit at lower frequency, in other myeloid malignancies, such as myelodysplastic syndromes (MDS; 3%–8%) and myeloproliferative neoplasms (MPNs; 2%–10%), as well as early T-cell precursor acute lymphoblastic leukemia (ETP-ALL; 16%–18%) (Brecqueville et al., 2011; Ewalt et al., 2011; Grossmann et al., 2013; Neumann et al., 2013; Stegelmann et al., 2011; Thol et al., 2011b; Traina et al., 2013; Walter et al., 2011). Similar to what is observed in de novo AML, R882 is most frequently targeted for mutation (60%) in these neoplasms (Thol et al., 2011b). Clinically, DNMT3A mutations also correlated with increased age and predicted prognosis in all types, including worse OS, event-free survival (EFS), and AML-free survival (Lin et al., 2011; Neumann et al., 2013; Renneville et al., 2012; Thol et al., 2011b; Walter et al., 2011). Paradoxically, univariate and multivariate analysis of 92 patients with MDS revealed DNMT3A mutations were correlated with better overall response rate (ORR) and progression-free survival (PFS) (Traina et al., 2013). Interestingly, similar to AML, MPNs showed an association between DNMT3A alterations and mutation in JAK2, IDH1/2, and ASXL1 but not TET2 (Stegelmann et al., 2011). However, studies in MDS and ETP-ALL have found no association between DNMT3A mutations and other known leukemogenic drivers, including FLT3 (Neumann et al., 2013; Thol et al., 2011b).
DNMT3A is a 102 kDa protein with three highly conserved functional domains: An N-terminal PWWP domain, a cysteine-rich PHD zinc finger domain, and a C-terminal catalytic domain (Hermann et al., 2004). DNMT3A mutations in cancer have been reported in all three domains, with most occurring in the catalytic domain, including the R882 mutation (60%) (Ley et al., 2010). Still, many cancer-specific mutations occur in non-catalytic domains. The PWWP domain is essential in localizing DNMT3A to heterochromatic regions of DNA during interphase, though it is unclear if this is related to its reported ability to bind DNA directly (Bachman et al., 2001; Ge et al., 2004; Purdy et al., 2010; Suetake et al., 2011). Alternatively, the PHD domain has also been shown to mediate regional specificity and repression through its interactions with transcriptional repressor RP58, HP1beta, histone deacetylatases (HDACs), and SUV39H1 (Datta et al., 2003; Fuks et al., 2001; Fuks et al., 2003). Exactly how these mutations disrupt protein function is of great interest, although a single unifying mechanism is unlikely to exist. Likewise, a recent clinical study found that R882 mutations confer poor prognosis in older populations, whereas non-R882 mutations confer poor prognosis in younger patients (Marcucci et al., 2012).
Whether these reported mutations are loss-of-function, gain-of-function, or act via a dominant-negative mechanism has also been debated. A strong case can be made that DNMT3A is an oncogene, as it is overexpressed in several cancers, depletion results in decreased proliferation and metastasis, and 5-aza-dC causes apoptosis through direct inhibition of DNMT3A (Oka et al., 2005). In regards to mutations themselves, the R882H DNMT3A mutant was sufficient to promote tumorigenesis in an IL-3 dependent transformation assay in leukemic 32D cells (Yan et al., 2011). Alternatively, some studies have demonstrated that mutant DNMT3A (R882H) has a markedly reduced catalytic ability (~50%) in methyltransferase assays and decreased DNA-binding capacity in vitro, implying a possible loss-of-function phenotype via a dominant-negative mechanism (Jia et al., 2007; Ley et al., 2010; Yamashita et al., 2010). Clearly, additional studies are necessary to understand the exact nature by which these cancer-associated mutations are transforming.
Characterizing larger DNMT3A-induced changes in the cancer methylome has proven quite challenging. Ley and colleagues showed no changes in genome-wide methylation according to DNMT3A status in AML, and although 182 specific regions showed increased hypomethylation in mutant samples, this did not correlate with gene expression (Ley et al., 2010). However, in a more recent cohort, a total of 3878 genomic regions were found to have significantly different methylation patterns using MeDIP-chip and differences of expression levels in 889 of 20,723 annotated genes was observed via an Affymetrix microarray (Yan et al., 2011). Further, this group found during RT-PCR validation that the expression of several HOX family genes significantly increased in DNMT3A mutant samples compared to wild-type.
Recent experiments may offer some insight behind the conflicting methylation and functional data. Protein binding at the DNMT3A tetramerization interface is important for methylation patterning, inducing processive methylation of clustered sites (Jia et al., 2007). Most mutations, including R882, occur within this tetramer interface. Therefore, differences between oligomerization states can explain how DNMT3A mutations alter epigenetic silencing and lead to transformation, without global changes in DNA methylation (Holz-Schietinger et al., 2011; Holz-Schietinger et al., 2012). Although the commonly occurring R882H mutation does not disrupt DNMT3A association with required cofactor DNMT3L in vitro, the latter is only expressed in early development (Chedin et al., 2002; Jia et al., 2007; Webster et al., 2005; Yamashita et al., 2010). However, other binding proteins, such as thymine-DNA glycosylase and ecotropic viral intergration site 1, can adhere to sites within the DNMT3A catalytic domain and may explain altered mutant DNMT3A activity (Li et al., 2007; Senyuk et al., 2011). In addition, non-catalytic mutations may disrupt protein binding to other domains, as described above. Although the exact mechanism remains elusive, these mutations result in decreased methylation processivity and altered localization, possibly to euchromatic regions of DNA (Jurkowska et al., 2011). This may help explain how DNMT3A mutations drive tumor formation in hematopoietic malignancies, even in the absence of larger global methylation changes.
TET2
The ten-eleven translocation (TET) family proteins were first discovered in cancer with the fusion of TET1 to MLL in select AML patients with t(10;11)(q22;q23) (Lorsbach et al., 2003). Mechanistic studies then showed the TET proteins are dioxygenases that depend on 2-oxoglutarate, oxygen, Fe(II), and ascorbic acid to catalyze the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC) at CpG regions in DNA (Blaschke et al., 2013; Ito et al., 2010; Minor et al., 2013; Tahiliani et al., 2009; Yin et al., 2013). TET enzymes may be responsible for DNA demethylation through both passive and active means. For example, CpG dinucleotides that are “marked” with 5-hmC are not recognized by DNMT1 and therefore, methylation is passively lost at these sites through repeated cycles of cell division (Valinluck and Sowers, 2007). Alternatively, active demethylation can proceed following placement of 5-hmC via the activation-induced cytidine deaminase (AID)-APOBEC DNA repair pathway (Guo et al., 2011). More recently, an even greater role for TET enzymes in active demethylation was shown in vitro, with TET enzymes proving sufficient for converting 5-mC to 5-hmC, 5-hmC to 5-formylcytosine (5-fC), and finally 5-fC to 5-carboxylcytosine (5-caC). 5-caC is then targeted by base excision repair enzymes to complete the demethylation process (He et al., 2011; Ito et al., 2011).
TET2 was first suspected to have a role in cancer when six patients with either secondary AML (sAML) or MDS were noted to have minimal deletions via FISH on chromosome 4q24 (Viguie et al., 2005). Soon after, the first TET2 somatic mutations were identified in 25 patients (14%) with MPNs (Delhommeau et al., 2008). Delhommeau and colleages then sequenced TET2 in patient tumor samples, becoming the first group to identify TET2 mutations in multiple myeloid neoplasms, including MDS (19%), MPNs (12%–14%), and sAML (24%). They concluded that TET2 was a novel bona fide tumor suppressor, noting that the majority of mutations are heterozygous (55%) and that TET2 defects precede the well-known JAK2 V617F driver mutation in MPN HSCs (Delhommeau et al., 2009). More recently, Schaub and colleagues disputed this result using colony formation assays to show that TET2 mutations can either precede (4 of 8 patients), follow (2 of 8), or occur independently (2 of 8) of JAK2 V617F mutations in MPN patient samples (Schaub et al., 2010). Although the temporal relationship between TET2 mutations and other leukemogenic drivers is still unclear, the frequency and ubiquitous nature of these mutations in cancer is quite revealing. In addition to MDS, sAML, and MPNs, TET2 mutations have now been described in other myeloid neoplasms, such as de novo AML (12%) and chronic myelomonocytic leukemia (CMML; 42%–46%) (Abdel-Wahab et al., 2009; Smith et al., 2010).
Unlike DNMT3A mutations, TET2 alterations seem to hold limited prognostic utility in leukemia. The overwhelming majority of studies published to date have found no change in OS or any other prognostic tools between patients harboring TET2 mutations and those who are not. However, one study in a cohort of 96 MDS patients reported that TET2 mutations conferred an OS, EFS, and AML-free survival advantage (Kosmider et al., 2009). Paradoxically, Abdel-Wahab and colleagues found that TET2 mutations were linked to worse OS in 93 patients with de novo AML, and a recent whole-exome sequencing study reported worse EFS in AML patients with TET2 mutation (Abdel-Wahab et al., 2009; Weissmann et al., 2012). Further, Nibourel and colleagues reported no association with OS or DFS in their cohort of de novo AML patients, though the prevalence of TET2 mutations in patients who failed to achieve complete remission (CR) trended higher (27% vs. 17%) (Nibourel et al., 2010). Despite questionable association to patient outcomes, TET2 mutations are linked with other clinical features, including monocytosis, leukocytosis, and advanced age at diagnosis (Jankowska et al., 2009; Smith et al., 2010; Tefferi et al., 2009a; Tefferi et al., 2009c). Although TET2 mutations show little association to known myeloid leukemogenic drivers—FLT3-ITD, RUNX1, CEBPA—they do associate with NPM1 and ASXL1 mutations and infrequently co-occur with IDH1 or IDH2 mutations (Chou et al., 2011a; Weissmann et al., 2012). Lastly, despite some data indicating no association between cytogenetics and TET2 status in MPN, TET2 mutations occur more often in the presence of normal karyotype and intermediate-risk AML (Hussein et al., 2010; Weissmann et al., 2012). In this cytogenetic setting, TET2 mutations do predict significantly worse OS in AML (Chou et al., 2011a).
Although few TET2 recurrent mutations have been reported, many mutations result in a frameshift or early stop codon and are therefore inactivating (Tefferi et al., 2009b). In fact, the largest proportion of nonsense mutations occur in exon 3, resulting in a truncated protein lacking the C-terminal catalytic domain (Moran-Crusio et al., 2011). Additionally, several missense mutations have been characterized as loss-of-function, with Ko and colleagues reporting impaired hydroxylation of 5-mC when mutant TET2 was overexpressed in HEK-293T cells. Furthermore, TET2 mutation status is significantly correlated with decreased global 5-hmC in myeloid tumors (Ko et al., 2010; Konstandin et al., 2011). Functional studies manipulating TET2 have also been able to recapitulate phenotypes that are characteristic of myeloid neoplasms, suggesting that TET2 loss may be a key event in leukemic transformation. For example, a conditional mouse model for TET2 loss in the hematopoietic compartment resulted in increased HSC self-renewal and myeloproliferation including splenomegaly, monocytosis, and extramedullary hematopoiesis (Moran-Crusio et al., 2011). This is consistent with other studies showing that TET2 inactivation leads to decreased 5-hmC in HSCs, amplification of the stem cell population, and may skew HSCs toward a myeloid lineage (Pronier et al., 2011; Quivoron et al., 2011).
The true effect of TET2 mutations on DNA methylation status has been difficult to ascertain. Despite an expected increase in 5-mC following TET2 inactivation, several studies have reported a global decrease in methylation (Ko et al., 2010; Yamazaki et al., 2012). However, analysis of specific gene promoters shows mixed results in TET2 mutant samples, frequently exhibiting promoter-specific hypermethylation in spite of global hypomethylation (Perez et al., 2012; Wu et al., 2011; Yamazaki et al., 2012). Still, Ko and colleagues noted that several AML patients with wild-type TET2 had 5-hmC levels very similar to those patients with mutant TET2 (Ko et al., 2010). This suggests a more complex relationship between TET2, DNA methylation status, and malignant transformation.
In the past year, exciting new evidence has emerged to suggest a more diverse role for TET2 in epigenetic regulation. In addition to known associations with polycomb repressive complex (PRC) regulator SIN3A and NURD complex member MBD3 (Wu et al., 2011; Yildirim et al., 2011), TET2 was recently identified as a direct binding partner with O-linked beta-N-acetylglucosamine transferase (OGT), an enzyme that marks histone H2B S112 at active transcription start sites (TSS) (Chen et al., 2013b). Although OGT doesn’t influence TET2 activity in functional assays, TET2 seems to actively target OGT to unmethylated promoters and activate transcription via other means (Vella et al., 2013). Furthermore, Deplus and colleagues showed that TET2 and OGT co-localize at active promoters marked by H3K4me3 through a direct interaction with host-cell factor 1 (HCF1) and that knockdown of TET2 leads to global decreases of H3K4me3 and GlcNAcylation (Deplus et al., 2013). Another direct interaction has been described between TET2 and EBF1, a transcription factor that is associated with transcriptional activation and “poised” chromatin (Guilhamon et al., 2013; Treiber et al., 2010). Additional binding partners have also been reported within the past year, including NANOG and IDAX (Costa et al., 2013; Ko et al., 2013). Lastly, several novel miRNAs were discovered to negatively regulate TET2 expression, offering a possible explanation for TET-associated transformation in the absence of any genomic alterations (Cheng et al., 2013; Fu et al., 2013; Song et al., 2013). Collectively, evidence is mounting that TET2 inactivation in cancer may alter more than just DNA methylation; in fact, transformation may result considerably from disrupted interactions with other epigenetic regulators and development-associated transcription factors.
IDH1/2
Isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) are two homodimeric metabolic enzymes that convert isocitrate to α-ketoglutarate (α-KG) while reducing NADP+ to NADPH. IDH1 is present in the cytosol and peroxisomes whereas IDH2 is located exclusively in mitochondria (Geisbrecht and Gould, 1999; Winkler et al., 1986; Xu et al., 2004). Frequent recurrent mutations in IDH1 were initially discovered in GBM (12%) following whole-exome sequencing of 22 patient tumor samples (Parsons et al., 2008). Further sequencing efforts revealed that mutations are most prevalent in WHO grade II/III gliomas (71%) and secondary GBMs (88%) but less common in primary GBMs (7%) (Balss et al., 2008). Subsequent studies have shown that IDH2 mutations are also enriched in WHO grade II/III gliomas, albeit less frequently, and that IDH1/2 mutations occur in a mutually exclusive manner (Hartmann et al., 2009). These data indicate IDH mutation is an early event in glioma oncogenesis, frequently preceding known alterations like TP53 mutation and 1p/19q loss (Watanabe et al., 2009). Interestingly, recent data suggest IDH1 and IDH2 mutations may actually differentially associate with astrocytoma and oligodendrogliomas, respectively (Hartmann et al., 2009). IDH mutations are associated with MGMT promoter hypermethylation, TP53 mutation, 1p/19q codeletion, ATRX inactivation, younger age, and improved prognosis while being inversely correlated with EGFR amplification in glioma (Chou et al., 2010; Wiestler et al., 2013; Yan et al., 2009; Zou et al., 2013). Further, although early studies could not find any IDH mutations in other types of solid tumors (Bleeker et al., 2009; Kang et al., 2009), recurrent mutations have since been identified in chondrosarcoma (56%), cholangiocarcinoma (23%), melanoma (10%), and prostate cancer (2%) (Amary et al., 2011; Borger et al., 2012; Ghiam et al., 2012; Shibata et al., 2011).
Soon after the discovery of IDH mutations in glioma, recurrent mutations of IDH were also identified in AML (Green and Beer, 2010; Mardis et al., 2009). Similar to glioma, IDH1 and IDH2 mutations are mutually exclusive, though the mutational frequency of IDH in AML is much lower (23%) (Chou et al., 2011b; Ward et al., 2010). In contrast, the utility of IDH mutation status as an independent prognostic marker in AML is less clear. In a convincing cohort of 493 adult patients with AML, Chou and colleagues found that IDH1 mutation had no impact on OS (Chou et al., 2010). Still, other studies have suggested a more disparate role, with mutation in IDH1 and IDH2 conferring poor and improved prognosis, respectively (Chou et al., 2011b; Schnittger et al., 2010). Notably, although another large cohort of 805 patients found that IDH mutation did not correlate with prognosis, a subset of patients with IDH mutation and CN-AML, NPM1 mutation, and no FLT3-ITD did show significantly reduced OS and RFS (Paschka et al., 2010). Therefore, it is likely that the use of prognostic subsets—IDH status along with other genetic markers—may improve the utility of IDH status as a biomarker in AML. Other features that correlate with IDH mutation status include normal karyotype, intermediate-risk cytogenetics, NPM1 mutation, and M1 AML subtype (Chou et al., 2010; Schnittger et al., 2010).
To date, practically all IDH mutations found in cancer are heterozygous and highly recurrent. Amino acid substitutions at residues IDH1-R132, IDH2-R172 and IDH2-R140Q are the most common, with considerable variability at R132 (R/H, R/C, R/S, R/G, R/L, R/V) and R172 (R/G, R/M, R/K) (Balss et al., 2008; Yan et al., 2009). The remarkable absence of any frameshift or nonsense mutations, deletions, or epigenetic silencing provided early evidence that IDH mutations were activating (Flanagan et al., 2012; Zhao et al., 2009). The R132 residue is located in the active site of IDH1 where it forms 2 hydrogen bonds with α- and β- carboxylate of isocitrate, its substrate (Nekrutenko et al., 1998; Xu et al., 2004). Initially, it was believed that these mutations may be loss-of-function or dominant-negative, as mutant IDH showed a reduced affinity for isocitrate and produced markedly less α-KG and NADPH in vitro (Yan et al., 2009; Zhao et al., 2009). However, an in vitro metabolite screen revealed that IDH mutations are neomorphic, producing the novel oncometabolite 2-hydroxyglutarate (2-HG) through heterodimer formation with the remaining wild-type IDH1. This was also verified in patient samples, with a strong correlation between the amount of 2-HG in tumor tissue and IDH1/2 mutation status (Dang et al., 2009; Ward et al., 2010). In fact, 2-HG levels are increased 10–100 fold in patient sera and can be used to reliably diagnose IDH status and monitor response to therapy, though this application may be restricted to myeloid neoplasms (Capper et al., 2012; DiNardo et al., 2013; Ward et al., 2010).
The effects of mutant IDH are pleiotropic and affect numerous cell processes including DNA methylation, histone methylation, HIF1a signaling, collagen synthesis, and metabolism (Cairns and Mak, 2013). Remarkably, α-KG levels are unchanged in mutant IDH AML and glioma (Dang et al., 2009; Gross et al., 2010), and it is now clear that 2-HG-mediated inhibition of 2-OG-dependent dioxygenases is the dominant mechanism by which IDH mutations are oncogenic (Xu et al., 2011). Early data from the Cancer Genome Atlas project first identified the glioma hypermethylator phenotype (G-CIMP) in GBM and its association with IDH mutations (Laffaire et al., 2011; Noushmehr et al., 2010). Following this, Figueroa and colleagues demonstrated that 2-HG inhibition of the α-KG-dependent enzyme TET2 actively generates the hypermethylator phenotype in AML. Further, they showed that TET2 and IDH mutations are mutually exclusive in AML, result in overlapping methylation signatures, and impair HSC differentiation in 32D myeloid cells (Figueroa et al., 2010). Similarly, work from our lab demonstrated that IDH1 mutation directly causes the G-CIMP phenotype, reduces global 5-hmC through TET2 inhibition, results in hypermethylation of the repressive histone marks H3K9 and H3K27, and blocks differentiation (Turcan et al., 2012). It is important to highlight that widespread loss of 5-hmC is an additional epigenetic hallmark in IDH or TET2 mutated cancers, including melanoma, and that reestablishment of the 5-hmC landscape can suppress tumor invasion and growth in both melanoma cells and a zebrafish model (Lian et al., 2012). Interestingly, IDH-associated increases in histone methylation are likely due to 2-HG-mediated inhibition of the Jumonji C (JmjC)-domain-containing histone demethylases (Lu et al., 2012). Still, others have proposed additional mechanisms in IDH-mutated cancers such as HIF1a stabilization through PHD inhibition, altered ECM structure due to decreased hydroxylation of collagen, and possible metabolic shifts in NADP/NADPH ratio (Sasaki et al., 2012; Zhao et al., 2009).
Recently, several exciting studies have shed light on novel mechanisms by which IDH mutations initiate malignant transformation and how underlying mechanisms may be exploited for therapeutic gain. In mouse models of leukemia and melanoma, IDH mutants accelerated cell cycle transition by activation of the MAPK/ERK pathway and repression of tumor suppressors CDKN2A and CDKN2B (Chaturvedi et al., 2013; Shibata et al., 2011). Although several studies have noted that IDH mutations cause increased colony formation in soft agar and enhanced proliferation, two mouse models for leukemia found that IDH mutation primes cells by inducing an MDS- or MPN-like state. However, combining IDH1 mutants with HOXA9, or IDH2 mutants with FLT3 or NRAS, was sufficient to initiate transformation (Chaturvedi et al., 2013; Chen et al., 2013a; Xu et al., 2011). This may be cancer-specific though, as mutant IDH2 alone was recently shown to be sufficient to induce sarcoma formation in mice, at least in one model system (Lu et al., 2013). Regardless, the primary mechanism underlying IDH-induced oncogenesis in several model systems is a block in cell differentiation (Pirozzi et al., 2013). Both groups showed restoration of differentiation and increased apoptosis following treatment with IDH inhibitor HMS-101 or Brd4 inhibitor JQ1. Similarly, Losman and colleagues showed that IDH mutant leukemic transformation is specific to the (R)-enantiomer of 2-HG, which can independently promote cytokine independence and block differentiation. Again, this transformation was reversible with IDH inhibitor AGI-5198 (Losman et al., 2013). Lastly, IDH inhibition can reverse novel EMT-associated expression patterns, though a lengthy delay to phenotypic change suggests more stable epigenetic alterations may be to blame (Grassian et al., 2012).
Despite the targeted nature of IDH inhibitors, IDH mutation likely unleashes epigenetic marks that are themselves selectable, such as DNA methylation. There is thus interest in the therapeutic potential of 5-azacytidine (5-aza) and decitabine (DAC), due to the complex downstream effects of IDH mutations on the cancer methylome. In order to determine if broader epigenetic therapies could “unlock” glioma initiating cells (GIC) from a dedifferentiated state, we treated both wild-type and mutant IDH cell lines with DAC and/or IDH inhibitor AGI-5198. Along with Borodovsky and colleagues, our group found that hypomethylating agents potently induce differentiation, impair colony formation, and suppress in vivo growth in IDH mutant cells only (Borodovsky et al., 2013; Turcan et al., 2013). This demonstrates that mutant IDH-induced DNA methylation likely plays a role in maintaining the self-renewal capacity of glioma tumor initiating cells. Similar effectiveness has also been seen in leukemia and chondrosarcoma (Chaturvedi et al., 2013; Chen et al., 2013a; Lu et al., 2013). Interestingly, IDH inhibition using the mutant IDH1 inhibitor AGI-5198 was not nearly as effective, which suggests that broader epigenetic therapies may be necessary to reverse more permanent changes induced by long exposure to mutant IDH. In addition, combination therapy with other IDH affected processes such as histone hypermethylation may have a role and warrant further investigation.
HISTONE METHYLATION
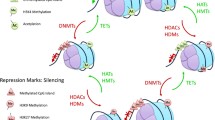
Histone methylation is a reversible process that takes place at the amino acid side chains of lysine, arginine, and histidine residues. Lysine methylation on histones H3 and H4 is the best characterized and catalyzed by the lysine methyltransferases (KMTs) through the required methyl group donor SAM. All of the KMTs except DOTL1/KMT4 have a catalytically active SET domain and are highly specific to both histone residue and degree of methylation (mono- vs. di- vs. tri-methylation) (Feng et al., 2002; Rea et al., 2000). Generally, methylation at H3K4, H3K36, and H3K79 corresponds to euchromatic or transcriptionally active regions of the genome, whereas methylation at H3K9, H3K27, and H4K20, is associated with heterochromatic regions and gene silencing. In addition, each residue is capable of four methylated states: unmethylated or mono-/di-/tri-methylated. This provides further regulatory diversity in the histone code. For example, H3K4me2/3 is found at TSSs of active genes, whereas H3K4me1 tends to localize to enhancer regions (Greer and Shi, 2012; Kampranis and Tsichlis, 2009).
In contrast, the lysine-specific demethylases (KDMs) work in opposition to the KMTs through the catalytic removal of methylation marks on histone tails. The two families of KDMs responsible are the (FAD)-dependent amine oxidases and the larger JmjC-containing family of α-KG/Fe(II)-ion dependent oxygenases (Shi and Whetstine, 2007). KDM1A/B (LSD1/2) and KDM5A-D (JARID1A-D) catalyze demethylation at H3K4, whereas KDM2A/B (JHDM1A/B) and KDM4A-C (JMJD2A-C) target H3K36, leading to repressed gene transcription at these sites. Alternatively, transcriptional activation is induced in part by demethylation at H3K9 and H3K27 by KDM1A, KDM3A-C (JHDM2A-C), or KDM4A-D and KDM6A/B (UTX/JMJD3), respectively (Varier and Timmers, 2011). Due to the broad and essential nature of these epigenetic marks across the genome, genetic aberrations of histone modifiers have powerful effects on vital cellular processes such as differentiation and cell cycle control, among others.
Writers (KMTs)
EZH2
EZH2/KMT6 is the enzymatic component of the polycomb repressor complex 2 (PRC2), which is responsible for methylation at H3K27 and subsequent gene silencing (Kirmizis et al., 2004). Other essential subunits of the PRC2 complex through which EZH2 interacts include embryonic ectoderm development (EED), suppressor of zeste 12 homologue (SUZ12), and RBAP48/RBBP4. Collectively, these polycomb group (PcG) proteins have been shown to regulate vital cellular processes including differentiation, cell identity, stem-cell plasticity, and proliferation (Margueron and Reinberg, 2011; Shih et al., 2012). As a result, aberrations in any PRC2 component can have powerful physiologic consequences on the cell.
Alterations in EZH2 were first discovered in breast and prostate cancer, where amplification and overexpression first implied it may function as an oncogene (Kleer et al., 2003; Varambally et al., 2002; Yang and Yu, 2013). This finding was validated both in vitro and in vivo, with EZH2 overexpression proving sufficient to drive proliferation in cancer cells and transform primary fibroblasts (Bracken et al., 2003; Croonquist and Van Ness, 2005). Recent sequencing studies have identified numerous mutations of EZH2 in a variety of leukemias and lymphomas, including follicular lymphoma (FL; 7%–22%), diffuse large B-cell lymphoma (DLBCL; 14%–21.7%), high grade B-cell lymphoma (18%), MDS/MPN (6%–13%), CMML (11.1%), T-ALL, and AML (Abdel-Wahab et al., 2011; Bodor et al., 2011; Capello et al., 2011; Ernst et al., 2010; Grossmann et al., 2011; Lohr et al., 2012; Makishima et al., 2010; Morin et al., 2010; Nikoloski et al., 2010; Ryan et al., 2011; Zhang et al., 2012). Interestingly, frequent missense and truncating mutations were observed, which generated some confusion in the field about whether EZH2 could possess both pro- and anti-oncogenic functions. Clinically, EZH2 mutations seem to commonly predict poor prognosis—worse OS/leukemia-free survival (LFS), high-risk IPSS score—especially in myeloid malignancies (Guglielmelli et al., 2011; Khan et al., 2013; Nikoloski et al., 2010). Additionally, EZH2 mutation associates with BCL2 rearrangement in FL and germinal center B-cell like DLBCL (GCB-DLBCL) and is notably absent from activated B-cell like DLBCL (ABC-DLBCL) (Beguelin et al., 2013; Morin et al., 2010; Ryan et al., 2011).
One landmark finding that emerged from the wealth of recent sequencing data was the identification of the highly recurrent heterozygous Y641 mutation (Y/F, Y/N, Y/H, Y/C) in FL, DLBCL, and other lymphoid neoplasms (Bodor et al., 2013; Bodor et al., 2011; Morin et al., 2010). Initially thought to be inactivating due to reduced catalytic ability against a short H3-like peptide in vitro, Y641 mutant EZH2 exhibited a powerful gain-of-function phenotype when incubated against the entire nucleosomal unit (Sneeringer et al., 2010). In addition, Sneeringer and others have observed a powerful synergy between the wild-type and Y641 forms of EZH2 both in vitro and in vivo. Whereas EZH2 is very efficient at catalyzing monomethylation of H3K27 but not di-/tri-methylation, EZH2 Y641 shows enhanced ability for di-/tri- methylation at H3K27 (Ryan et al., 2011; Wigle et al., 2011; Yap et al., 2011). Similar findings have also been observed with A677G and A687V mutant EZH2, though these are far less prevalent in cancer (Majer et al., 2012; McCabe et al., 2012a). Since almost all EZH2 gain-of-function mutations are heterozygous, the overall consequence of these mutations is a cooperative and thereby efficient silencing of genes associated with the repressive H3K27 mark.
Recent studies have shown that mutant EZH2-driven tumors can be effectively targeted with small molecule inhibitors. Knutson and colleagues were the first to describe potent phenotypic effects in lymphoma cell lines, following treatment with the SAM-competitive EZH2 inhibitor (EPZ005687) (Knutson et al., 2012). This inhibitor was highly selective, inducing cell death in mutant EZH2-expressing cells only. As expected, these cells showed global reduction of the H3K27me2/me3 histone mark and significant enrichment of cell cycle gene sets by GSEA. The more recent EZH2 inhibitor GSK126 was also highly selective for mutant EZH2 lymphoma cells in vivo and led to increased activation of known EZH2 target genes, such as TXNIP and TNFRSF21 (McCabe et al., 2012b). Although EZH2 mutation alone may be insufficient to induce development of B-cell lymphoma, new evidence suggests it functions as a master regulator of GCB phenotype through repression of CDKN1A, IRF4, and PRDM1 (Beguelin et al., 2013). Due to frequent activation of EZH2 in lymphoma, these new targeted therapies hold exciting promise in the clinic.
SETD2
The major KMT responsible for H3K36 trimethylation is SETD2/KMT3A, which is a novel candidate tumor suppressor gene (TSG) (Edmunds et al., 2008). Gene deletions in clear cell renal cell carcinoma (ccRCC)-derived cell lines are common, reduced expression is seen in breast tumors, and loss is associated with decreased H3K36 trimethylation (Duns et al., 2012; Duns et al., 2010; Newbold and Mokbel, 2010). SETD2 mutations are quite common in ccRCC (7.4%–11.6%), pediatric high-grade glioma (HGG; 15%), and adult HGG (8%) (Cancer Genome Atlas Research Network, 2013; Dalgliesh et al., 2010; Duns et al., 2010; Fontebasso et al., 2013; Hakimi et al., 2013b; Varela et al., 2011). Almost all of the mutations characterized so far are frameshift or nonsense and therefore truncating, further suggesting SETD2 may be an important TSG in select malignancies (Hakimi et al., 2013a). Furthermore, Hakimi and colleagues found that SETD2 mutations were significantly associated with worse cancer-specific survival (CSS) in ccRCC (Hakimi et al., 2013b). Though phenotypic effects of SETD2 inactivation have not been clarified, recent research showed that SETD2 loss triggers MSI and can increase genome-wide mutation rates through alterations in H3K36 methylation (Li et al., 2013a; Schmidt and Jackson, 2013).
MLLs
The mammalian mixed lineage leukemia (MLL) family of genes encodes a series of active (MLL1–4/KMT2A–D) and inactive (MLL5/KMT2E) KMTs, which have all been implicated in cancer. MLL1–4 are responsible for methylation at H3K4 and share a common core formed by WDR5, RbBP5, Dpy-30, and Ash2L (Varier and Timmers, 2011). Notably, MLL1–2 form a complex along with the menin (MEN1) tumor suppressor and recent evidence shows that H3K27 demethylase KDM6A/UTX can complex with MLL2–4 (Hughes et al., 2004; Yokoyama and Cleary, 2008; Yokoyama et al., 2005).
The earliest known alterations in MLL family genes involved frequent rearrangements of MLL1 at 11q23, with recombination involving more than 40 different partner genes and occurring in 60%–80% of infants with ALL or AML (Dimartino and Cleary, 1999; Pais et al., 2005; Thirman et al., 1993). Since then, several missense and truncating mutations have been identified in MLL1 in bladder, lung, and breast cancer (Gui et al., 2011; Kan et al., 2010). Although MLL1 acts as a dominant oncogene in liquid tumors, these new discoveries suggest a different recessive role for MLL1 in some solid tumors (Krivtsov and Armstrong, 2007). Infrequent truncating mutations have also been noted in MLL4 in medulloblastoma and head and neck squamous cell carcinoma (HNSCC), suggesting a minor but alternative role for this family member as well (Pugh et al., 2012; Stransky et al., 2011).
Notably, recently, many new mutations have been identified in both MLL2 and MLL3, showing diversity of both mutation and tumor type. MLL2 and MLL3 mutations are frequently nonsense or frameshift, resulting in a truncated protein lacking the active SET domain (Morin et al., 2011; Parsons et al., 2011; Pasqualucci et al., 2011b; Pugh et al., 2012; Stransky et al., 2011). Along with MLL1/4, MLL2/3 mutations suggest a dual role for MLL family proteins in oncogenesis, which may depend heavily on cellular context. In addition, mutations have been discovered in numerous cancers, occasionally at high frequency, including colon (MLL3, 14%–17%), DLBCL (MLL2, 24%–32%), FL (MLL2, 89%), AML, breast, GBM, RCC, prostate, pancreatic, bladder, medulloblastoma, and HNSCC (Balakrishnan et al., 2007; Dalgliesh et al., 2010; Gui et al., 2011; Li et al., 2013b; Lindberg et al., 2013b; Mann et al., 2012; Morin et al., 2011; Parsons et al., 2008; Parsons et al., 2011; Pasqualucci et al., 2011b; Pugh et al., 2012; Sjoblom et al., 2006; Stransky et al., 2011; Vakoc et al., 2009; Watanabe et al., 2011). Unfortunately, functional data remains sparse and the importance of these mutations has yet to be characterized. Watanabe and colleagues made the interesting observation that in colorectal carcinoma, MLL3 mutations were associated with increased MSI, though no mechanism has been proposed (Watanabe et al., 2011). Regardless, they are intriguing candidates for further study in cancer, especially since MLL family proteins are important regulators of HOX proteins and differentiation (Wang et al., 2009a).
NSD1/2
Histone-lysine N-methyltransferase NSD2/MMSET was first implicated in cancer as a target for rearrangement [t(4;14)(p16.3;q32)] in 15%–20% of multiple myeloma (MM) patients (Chesi et al., 1998). This translocation results in aberrant upregulation of NSD2, which first suggested that it may be an oncogene. Subsequent work has shown that knockdown in MM KMS11 cells leads to apoptosis and re-expression of wild-type NSD2 causes increased proliferation (Martinez-Garcia et al., 2011). Furthermore, overexpression of wild-type NSD2 is sufficient to transform NSD2-/- cancer cells in vivo and in mouse embryonic fibroblasts (MEFs) (Kuo et al., 2011). Functionally, interactions with HDAC1/2 and catalytic activity at H3K4 and H4K20 have been proposed, though these may be minor (Marango et al., 2008). It is now clear that the NSD2-catalyzed conversion of unmethylated H3K36 to mono- or di-methylated forms, with concomitant decreases in H3K27me3, is the dominant mechanism driving NSD2-associated oncogenic reprogramming (Kuo et al., 2011; Li et al., 2009). In fact, Kuo and colleagues demonstrated that NSD2 SET catalytic activity is required for transcriptional activation at several oncogenic loci (TGFA, MET, PAK1, RRAS2). Pathway analyses have also identified the following as significantly altered in mutant NSD2 tumors: TP53 pathway, cell cycle, DNA repair, focal adhesion, and Wnt (Kuo et al., 2011; Martinez-Garcia et al., 2011).
Recently, sequencing projects revealed the presence of a highly recurrent mutation in NSD2 (E1099K), which is present in 7.5% of pediatric B-ALL and other lymphoid neoplasms (Jaffe et al., 2013; Oyer et al., 2013). These studies showed that NSD2 E1099K leads to enhanced colony formation in soft agar and expected increases and decreases in H3K36me2 and H3K27me3, respectively. This new discovery has exciting therapeutic potential—similar to the activating mutations in EZH2 described above. Additionally, the related KMT NSD1 was recently discovered to harbor point mutations in multiple cancers, including HNSCC and AML (Dolnik et al., 2012; Yan et al., 2011). If these mutations prove to be similarly activating, both NSD members will represent completely novel areas of epigenetic regulation through which small molecule targeted inhibition could be useful.
Erasers (KDMs)
KDM6A
Among the first cancer-associated mutations in KDMs that were identified were those in KDM6A following sequencing of 1,390 patient tumor samples (van Haaften et al., 2009). Remarkably, KDM6A mutations were found to be widespread across both solid and liquid tumors, including AML, chronic myelogenous leukemia (CML), T-ALL, MM, Hodgkin’s lymphoma (HL), TCC, breast, colon, esophageal, pancreas, endometrial, GBM, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), and RCC (Dalgliesh et al., 2010; Gui et al., 2011; Mann et al., 2012; Mar et al., 2012; Ross et al., 2013; van Haaften et al., 2009). Since then, KDM6A mutations have also been discovered in other tumors such as prostate cancer, medulloblastoma, and adenoid cystic carcinoma (Ho et al., 2013; Lindberg et al., 2013a; Robinson et al., 2012). Although these mutations occur at low frequency in most cancers, KDM6A mutations in bladder carcinoma are quite common (20%–29%) (Gui et al., 2011; Poon et al., 2013; Ross et al., 2013). Furthermore, KDM6A mutations in bladder carcinoma associate with earlier grade and are inversely correlated with stage (Gui et al., 2011). Therefore, KDM6A inactivation may be a powerful driver and early event in bladder oncogenesis. However, whether KDM6A mutation status holds significant prognostic value in cancer is yet to be determined.
Despite infrequent inactivation in many cancers, mutation and functional data have established that KDM6A is a bona fide tumor suppressor gene. KDM6A is a 1401 amino acid protein with several N-terminal tetratricopeptide-repeat (TPR) domains and a single C-terminal Jumonji C (JmjC) domain (Shpargel et al., 2012). Early sequencing efforts revealed that a majority of KDM6A mutations are either frameshift or nonsense, and since most occur before the active JmjC demethylase domain, they are most likely inactivating. To test this hypothesis, van Haaften and colleagues re-expressed wild-type KDM6A in KDM6A-deleted cell lines and observed markedly reduced proliferation (van Haaften et al., 2009). Similarly, we recently showed that cancer-specific missense mutations in the JmjC domain can abrogate this growth suppressive effect and may even contribute to a dominant proliferative phenotype. Furthermore, following overexpression, these tumor-specific mutants exhibited reduced demethylase activity at the repressive chromatin mark H3K27me3 (Ho et al., 2013). Dysregulation of methylation at H3K27 may have important consequences in cancer, as demethylation of H3K27 at HOX genes is required for proper differentiation. Interestingly, it has also been reported that KDM6A binds directly to the HOXB1 locus and that its activation requires KDM6A catalytic activity (Christensen et al., 2007; Morales Torres et al., 2013). In addition to regulation through HOX gene targets, KDM6A catalytic activity also activates RB pathway genes through HBP1 to further influence differentiation and cell cycle control (Herz et al., 2010; Wang et al., 2010).
In addition to contributing to tumor suppression via its catalytic domain, KDM6A also has important demethylase-independent roles in cancer. A recent study found that conditional inactivation of KDM6A in a mouse model did not change global levels of H3K27me3, though it did contribute to a MDS-like phenotype and reduced migration of HSCs (Thieme et al., 2013). Furthermore, it is also known that KDM6A can bind to KMTs MLL2–4 to promote H3K4 methylation independent of its catalytic domain and that demethylase-inactive KDM6A is sufficient to induce differentiation (Cho et al., 2007; Issaeva et al., 2007; Lee et al., 2007; Morales Torres et al., 2013). In fact, Wang and colleagues mapped the chromatin occupancy of KDM6A, H3K4me2, and H3K27me3 in primary human fibroblasts and discovered that 62% of KDM6A target genes are enriched for univalent H3K4me2 (Wang et al., 2010). Therefore, KDM6A has at least two independent, yet complimentary, mechanisms for shaping the epigenetic landscape in cancer. Indeed, one study found that inactivating mutations in the catalytic JmjC domain caused increased growth yet also led to simultaneous increases and decreases in H3K27me3 and H3K4me1, respectively (Herz et al., 2010). As a result, it is likely that the KDM6A-associated phenotypes in cancer are diverse and are linked to the type and location of each driving mutation.
Other KDMs
Although KDM6A mutations are the most prevalent and best characterized among the KDMs, several others have been identified as significantly mutated across cancer, albeit at low frequency (Cerami et al., 2012; Gao et al., 2013; Parsons et al., 2011; Pasqualucci et al., 2011b). Uniquely, ccRCC harbors mutations in many of the KDMs, including KDM1A, KDM2B, KDM3A, KDM3B, KDM4A/B, and KDM5C (Dalgliesh et al., 2010; Hakimi et al., 2013a; Larkin et al., 2012; Shi et al., 2011). The natural function of these KDMs is still being determined, but these studies and others suggest KDM1A, KDM4A–C, and KDM5B are putative oncogenes, whereas KDM6A/B and KDM3B/C are tumor suppressors. Also, KDM2A/B and KDM5A seem likely to be both pro- and anti-oncogenic, depending on context (Rotili and Mai, 2011). More recently, Niu and colleagues provided the first in vivo evidence that KDM5C serves as a tumor suppressor following VHL loss in ccRCC and that cancer-specific mutations were inactivating. Furthermore, they demonstrated that HIF2a binds directly to KDM5C, targeting KDM5C to demethylate H3K4me3 at HIF-repressed gene loci (Niu et al., 2012). It will be exciting to determine if some of these mutations are true oncogenic drivers. Additionally, if gain-of-function mutants are identified in KDM oncoproteins, these may be prime candidates for existing KDM inhibitors or new targeted therapies (Rotili and Mai, 2011).
HISTONE ACETYLATION
Lysine residues on histone tails may also undergo another form of covalent modification through the addition of an acetyl functional group. This process uniquely results in the neutralization of charge normally associated with lysine residues, which weakens the electrostatic interaction between histones and negatively charged DNA. As a result, it is believed that histone acetylation primarily results in a more “open” chromatin configuration, serving as a “mark” of active gene transcription. Several ChIP-seq studies have now confirmed this, showing localization of acetylated histones at enhancers, promoters, and even throughout the transcribed region of active genes (Dawson and Kouzarides, 2012; Heintzman et al., 2007; Wang et al., 2008). In addition to altering the chromatin state directly, these specific histone “marks” further act to recruit other remodelers containing “reader” bromodomains and tandem plant homeodomain (PHD) fingers (Taverna et al., 2007).
The process of histone acetylation is carried out by the lysine acetyltransferase (KAT) enzymes, of which there are two major classes: Type-A, which are usually found in the nucleus and Type-B, which are cytoplasmic and act on free histones. Dynamic regulation of acetylation is also catalyzed by the histone deacetylase (HDAC) enzymes, which oppose the actions of KATs and remove acetyl groups from histone tails. Interestingly, these enzymes are capable of modifying other non-histone proteins—including p53, Rb, and MYC—and have additional roles as transcriptional cofactors, which has led to many challenges in determining their specific roles in cancer and other disease processes (Dawson and Kouzarides, 2012; Iyer et al., 2004).
Writers (CREBBP and EP300)
CREB-binding protein (CREBBP) and E1A binding protein p300 (EP300) are structurally distinct from other KATs and have unique broad substrate specificity, including the ability to acetylate all four histones in vitro. In fact, both proteins are highly conserved, with 75% similarity across their entire length and 63% homology at the amino-acid level (Iyer et al., 2004; Shiama, 1997). Not surprisingly, many functional similarities exist. Both proteins engage in several diverse functions, including chromatin remodeling via KAT activity, acetylation of association proteins (p53, Rb, E2F), and the ability to act as scaffolds for transcription factors and other transcriptional machinery (Bannister and Kouzarides, 1996; Gu and Roeder, 1997; Nakajima et al., 1997). Despite being some of the earliest epigenetic modifiers identified, their roles in both normal physiology and disease are just beginning to be appreciated.
Both CREBBP and EP300 have long been linked to cancer, though the specific roles they play have been harder to elucidate. Suspicion that CREBBP may be a tumor suppressor first arose in the mid-1990s, when heterozygous germline mutations were identified in the setting of Rubinstein-Taybi syndrome, a developmental disorder with an increased prevalence of cancer, including leukemia and lymphoma (Petrij et al., 1995). Around the same time, EP300 was first shown to bind to the E1A viral oncoprotein, suggesting it may also function as a tumor suppressor (Eckner et al., 1994). Soon after these discoveries, the first genetic alteration of CREBBP in cancer was identified in M4/M5 AML subtypes, albeit a rare t(8,16)(p11,p13) translocation that fuses the MOZ gene with the N-terminus of CREBBP (Borrow et al., 1996; Panagopoulos et al., 2001). Interestingly, reports of MOZ-EP300 translocations do exist, though these events may be exceedingly rare (Lai et al., 1985). Though a few early studies identified low frequency EP300 mutations in colorectal carcinoma, breast cancer, and gastric cancer, the full spectrum of mutational inactivation of CREBBP and EP300 would not be fully evident until the genomics era.
Genomic and exomic sequencing data from the past several years have revealed that CREBBP and EP300 inactivation via mutation is more widespread and frequent than previously thought. For example, CREBBP mutations have now been described in NHL (21%), DLBCL (29%), FL (32.6%), TCC (13%), ACC (7%), and relapsed ALL (18.3%), with EP300 mutations occurring slightly less frequently in NHL (7%), DLBCL (10%), FL (8.7%), TCC (13%), ACC, and relapsed ALL (Gui et al., 2011; Ho et al., 2013; Morin et al., 2011; Mullighan et al., 2011; Pasqualucci et al., 2011a). Additionally, CREBBP and EP300 are collectively mutated in up to 18% of SCLC (Peifer et al., 2012). Interestingly, mutations in both genes are mutually exclusive, suggesting functional equivalency, at least in part. Additionally, the majority of mutations are heterozygous, indicating that both genes most likely function as haploinsufficient tumor suppressors. In line with this, an earlier mouse study showed that heterozygous CREBBP loss led to increased neoplasia over wild-type mice (Kung et al., 2000). In almost all of these studies, mutations strongly clustered in the catalytic KAT domain, several of which exhibit reduced acetyltransferase ability in vitro at H3K18 and in the non-histone substrates Bcl6 and p53 (Mullighan et al., 2011; Pasqualucci et al., 2011a; Peifer et al., 2012). Though the targets of CREBBP/p300 are diverse, it seems likely that disruption of acetyltransferase ability can be a main contributor to tumor formation.
Despite the identification and initial functional characterization of these tumor-specific mutations, the physiologic consequences leading to oncogenesis remain obscure. For example, in SCLC, CREBBP/EP300 mutations do not lead to any obvious concerted shifts in gene expression, even though several reported mutations are inactivating in vitro (Peifer et al., 2012). Due to the diverse roles of CREBBP and p300, it is possible that some inactivating mutations exert phenotypic consequences through other non-epigenetic mechanisms. For example, a more recent discovery showed that both proteins exhibit acetyltransferase specificity for histones H3 and H4 at double-strand break (DSB) sites, facilitating the recruitment of the SWI/SNF chromatin remodeling complex (Ogiwara et al., 2011). Also, CREBBP KAT mutations favor constitutive activity of the Bcl6 oncogene over p53, which may be alone sufficient to promote tumorigenesis. Lastly, paradoxical roles for CREBBP/EP300 have been described recently. For example, EP300 is actually upregulated in melanoma cell lines, and inhibition of KAT function in vitro reduces melanoma tumor cell growth (Yan et al., 2013). Additional experiments will be necessary to help identify functional consequences of these mutations and in what specific contexts these two KATs function as tumor suppressors.
Erasers (HDACs)
In contrast to acetyltransferases, the 18 member HDAC family is responsible for the removal of acetyl groups from lysine residues on histone tails. Similar to KATs, HDACs have a wide range of protein targets, and are also known to deacetylate nonhistone substrates (Ellis et al., 2009). The HDAC family has four major classes, with class I (nucleus), II (nucleus and cytoplasm), and IV requiring the zinc ion for catalytic activity. Class III (sirtuins) are catalytically active in the absence of zinc and share almost no homology with the other HDACs (New et al., 2012). Notably, several HDACs have been implicated in cancer. Specifically, functional experiments have revealed that these HDACs are pro-oncogenic, with increased apoptosis and reduced proliferation (Class I) or reduced angiogenesis and cell migration (Class II) following specific HDAC knockdown (Ellis et al., 2009). Most importantly, this role is congruent with their acetyltransferase counterparts, the KATs, which have been characterized as tumor suppressors (see above).
In the past decade, much work has been done to develop targeted inhibitors of HDACs, since they are now established oncogenes in many cellular contexts. These HDAC inhibitors (HDACi) work to prevent histone deacetylation, facilitating a more open chromatin configuration and leading to increased gene transcription. Vorinostat, the first FDA approved HDACi, is already in use for select neoplasms, and other more selective HDACi show promising efficacy in early trials at reducing cell growth and increasing apoptosis (Witt et al., 2009). Despite the substantial therapeutic potential for HDACi, there is a need to determine biomarkers for treatment response and resistance.
Sequencing data from the past several years has identified inactivating mutations in several HDACs, which may influence the effectiveness of HDAC inhibitor therapy and predict overall response in patients (You and Jones, 2012). Specifically, mutations have now been reported in HDAC2 (colon), HDAC4 (breast), and HDAC9 (prostate) (Berger et al., 2011; Ropero et al., 2006; Sjoblom et al., 2006). Of these, the recurrent frameshift mutation of HDAC2 in exon 1 has been the most extensively characterized. This mutation is incredibly common in colon cancer (21%), resulting in a premature stop codon and loss of measurable HDAC2 expression in 83% of mutant tumors (Ropero et al., 2006). Further, HDAC2 mutations are enriched in MSI colon cancers (43%) (Hanigan et al., 2008). Importantly, in vitro work showed that HDAC2-deficient cells were resistant to HDACi via trichostatin A, exhibiting no hyperacetylation at target histones H3 and H4 and no reduction in proliferation compared to wild-type HDAC2-expressing cells (Ropero et al., 2006). Recent work has identified the pro-apoptotic gene APAF1 as a likely target for repression by HDAC2, providing a specific mechanism for both HDACi efficacy and resistance in HDAC2 mutant cells, where HDACi does not appreciably alter APAF1 levels (Hanigan et al., 2008). Lastly, although there is an important role for using HDAC mutation status as a predictor of HDACi treatment response, Ropero and colleagues have further shown that the HDAC2 mutation itself can cause changes in gene expression, actively leading to increased levels of multiple pro-tumorigenic proteins (Ropero et al., 2008).
CHROMATIN REMODELING
In addition to gene regulation via covalent histone tail modifications, the ATP-dependent chromatin remodelers also shape chromatin structure and thereby affect gene expression patterns. Several multi-unit effectors share this responsibility, including SWI/SNF, ISWI, INO80, SWR1, and NURD/Mi2/CDH complexes. In the past several years, protein components of the SWI/SNF complex have been found to be frequently inactivated in cancer, and subsequent work has solidified their status as bona fide epigenetic tumor suppressors (Wilson and Roberts, 2011).
SWI/SNF complex
The SWI/SNF complex consists of one or two mutually exclusive catalytic ATPases (SMARCA2/BRM or SMARCA4/BRG1), a group of conserved core subunits (SMARCB1/SNF5, SMARCC1/BAF155, SMARCC2/BAF170), and other variant subunits (Wilson and Roberts, 2011). Two important SWI/SNF complexes implicated in cancer are the BAF and PBAF complexes, which contain the mutually exclusive ARID1A or ARID1B subunits and PBRM1 or BRD7 subunits, respectively (Reisman et al., 2009; Wang et al., 2013). Collectively, SWI/SNF complexes remodel chromatin through the mobilization of nucleosomes both by sliding and by the ejection/insertion of histone octomers (Saha et al., 2006). Through these mechanisms, the SWI/SNF complexes have powerful effects on transcriptional regulation, serving an important role in development through the coordinate activation and repression of critical gene expression programs. Importantly, specificity is most likely achieved through the unique combinatorial assembly of the SWI/SNF complex, facilitated by the sheer size and diversity of the protein subunit repertoire (Wang et al., 1996).
ARIDs
The AT-rich interactive-containing domain (ARID) gene superfamily consists of seven members (ARID1–5), of which the following have now been implicated in cancer: ARID1A/BAF250a, ARID1B/250b, and ARID2/BAF200. Mutations in ARID1A are the most widely reported in the literature, with remarkable frequency, first reported in ovarian clear cell carcinoma (OCCC; 50%) and endometrioid carcinoma (30%) (Bosse et al., 2013; Jones et al., 2010; Wiegand et al., 2010). Mutations in other cancers exist, including medulloblastoma, breast, lung adenocarcinoma, ACC, hepatocellular carcinoma (HCC), gastric, pancreatic, and neuroblastoma (Fujimoto et al., 2012; Ho et al., 2013; Sausen et al., 2013; Wu and Roberts, 2013; Zang et al., 2012). Interestingly, the majority of mutations are heterozygous, truncating, and evenly spread along the protein, suggesting a possible role as a haploinsufficient tumor suppressor. Functional studies have confirmed this, noting increased proliferation and colony formation, impaired differentiation, and decreased apoptosis following partial ARID1A knockdown (Gao et al., 2008; Luo et al., 2008; Nagl et al., 2007; Zang et al., 2012). Correspondingly, re-expression of ARID1A decreases cell proliferation (Zang et al., 2012). In addition, a role in differentiation seems likely, though conflicting data on the specific consequences of ARID1A inactivation has been complicated through varying technical approaches and model systems (Wu and Roberts, 2013).
Little is currently known about how ARID1A inactivation leads to malignant transformation through SWI/SNF chromatin remodeling, though several intriguing possibilities exist. Both ARID1A and ARID1B provide unique and mutually exclusive specificities for SWI/SNF recruitment to chromatin (Wilson and Roberts, 2011). Interestingly, with ARID1A, this process is at least partially independent of its ARID domain, which binds DNA in a non-specific manner only (Dallas et al., 2000). Instead, ARID1A likely contributes to specific recruitment of SWI/SNF by binding transcription factors and transcriptional coactivator/corepressor complexes, including nuclear hormone receptors (Nie et al., 2000; Trotter and Archer, 2004). In fact, Inoue and colleagues showed that re-expression of ARID1A in a breast cancer cell line augments transcriptional activation through glucocorticoid receptors, estrogen receptor, and androgen receptor (Inoue et al., 2002). This specificity is likely due to the presence of several nuclear hormone receptor binding sites near the C-terminus of ARID1A (Nie et al., 2000). In light of this, the impressive prevalence of ARID1A mutations in hormone-responsive neoplasms (ovary, breast) is likely to be more than just coincidence.
The clinical utility of ARID1A mutation status is currently unclear, due to a limited number of sufficiently powered studies. However, Sausen and colleagues recently found that ARID1A or ARID1B mutations associate with worse OS in patients with neuroblastoma, and a previous breast cancer study noted that decreased ARID1A expression can confer worse prognosis (Mamo et al., 2012; Sausen et al., 2013). Several other intriguing observations have been noted, including a tendency toward co-occurrence between mutations in ARID1A and CTNNB1 (β-catenin) or PI3K-Akt pathway alterations, as well as a mutual exclusivity between ARID1A and TP53 mutations (Bosse et al., 2013). Whether ARID1A-associated phenotypes arise through additional epigenetic means, such as reported binding partners HDAC1/2, its ubiquitin ligase activity at H2B K120, or by increased MSI, remains to be determined (Bosse et al., 2013; Li et al., 2010).
Mutations in other ARID family members ARID1B and ARID2 have now been reported, though investigation into their clinical and functional relevance is in its infancy. ARID1B mutations occur at moderate frequency in neuroblastoma (7%) and HCC (6.7%), with sporadic mutations identified in breast, gastric, and pancreatic cancers too (Fujimoto et al., 2012; Sausen et al., 2013; Shain et al., 2012; Wang et al., 2011). These mutations are usually frameshift and hemizygous, suggesting it may be a tumor suppressor, like ARID1A. More intriguing has been the discovery of ARID2 mutations in HCC (5.8%–6.5%), with strong enrichment in HCV-associated HCC (14%) (Fujimoto et al., 2012; Zhao et al., 2011). ARID2 seems to mediate anti-proliferative signaling by binding to IFN-inducible promoters to remodel chromatin in response to interferon signaling. As such, mutations in ARID2 may render IFN-related immune processes incapacitated in the setting of HCV infection, leading to accelerated tumorigenesis. More recently, mutations in ARID2 were identified in NSCLC (7.3%), making this gene one of the most frequently mutated in this type of cancer (Manceau et al., 2013).
SMARCs
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin (SMARC) genes, also known as BRG1-associated factors, are among the most frequently altered and best characterized chromatin remodelers in cancer. These genes encode several SWI/SNF proteins including the catalytic ATPase subunits (either SMARCA2 or SMARCA4), a group of conserved core subunits (e.g. SMARCB1, SMARCC1, and SMARCC2), and variant subunits (e.g. SMARCE1) (Wilson and Roberts, 2011). Tumor-specific mutations in the following SMARCs are especially common: SMARCB1/SNF5, SMARCA2/BRM, and SMARCA4/BRG1 (Shain et al., 2012). Alterations in these critical chromatin remodelers have profound effects on vital processes, such as differentiation, cell proliferation, and metastasis.
SMARCB1 is inactivated via biallelic alterations—deletion and mutation (truncating, missense)—in nearly all rhabdoid tumors (RTs; 98%), an especially lethal cancer that predominantly affects young children (Sievert et al., 2009; Versteege et al., 1998). Recent exomic sequencing data has identified mutations in other tumors and pre-malignant lesions, such as sarcoma, gastric carcinoma, schwannomatosis, meningioma, chordoma, and hepatoblastoma (Christiaans et al., 2011; Hulsebos et al., 2007; Kim et al., 2013; Kreiger et al., 2009; Mobley et al., 2010; Trobaugh-Lotrario et al., 2009). Mouse models for SMARCB1 loss have verified a tumor suppressive role for this protein, with homozygous loss leading to frequent formation of lymphomas and RT-like tumors at only 11 weeks, which is notably faster than TP53-inactivated models of sarcoma (Guidi et al., 2001; Roberts et al., 2000; Wilson and Roberts, 2011). Interestingly, SMARCB1-deleted mice do not develop tumors in the absence of SWI/SNF ATPase SMARCA4 (Wang et al., 2009b), suggesting that oncogenesis following SMARCB1 inactivation is due to aberrant residual activity of SMARCA4-containing SWI/SNF complexes. Additionally, global gains of H3K27me3 following loss of SMARCB1 may directly contribute to tumorigenesis, as simultaneous EZH2 loss protects against transformation. In fact, EZH2 elevation occurs immediately following SMARCAB1 inactivation, and re-expression of SMARCB1 evicts PcG proteins from the CDKN2A tumor suppressor locus, activating transcription of this important tumor suppressor (Kia et al., 2008; Wilson et al., 2010). This evidence provides both a necessary mechanism and a possible therapeutic target (EZH2) in SMARCB1-inactivated tumors.
Further mechanistic insights have yielded many other potential therapeutic avenues in SMARCB1-mutated tumors. For example, SMARCB1 plays a critical role in cell cycle control. Mouse models have shown that coinactivation with Rb or p16 does not accelerate tumorigenesis, and simultaneous loss of cyclin D1 protects against tumor formation (Isakoff et al., 2005; Oruetxebarria et al., 2004; Tsikitis et al., 2005). In this vein, cyclin D1 inhibitors may have surprising effectiveness in these patients. Furthermore, direct binding to Myc has also been reported, along with increased Myc expression following SMARCB1 loss (Cheng et al., 1999; Tsikitis et al., 2005). Though the efficacy of synthetic lethal approaches in targeting Myc-driven tumorigenesis has yet to be established, these strategies may be worthwhile therapeutic candidates. In addition to cell cycle dysregulation, SMARCB1 inactivation leads to alterations in cell differentiation, specifically by increasing hedgehog signaling through the transcription factor Gli1 (Jagani et al., 2010). Thus, inhibitors of Gli1 provide a fourth potential target for therapy in SMARCB1-inactivated tumors. Lastly, increased RHOA activity and subsequent cell migration was observed following knockdown of SMARCB1, though therapeutic possibilities of this mechanism remain unexplored (Caramel et al., 2008).
SMARCA4, one of two SWI/SNF ATPase subunits, was first identified as a potential tumor suppressor in NSCLC, where expression is lost in 15%–50% of all tumors (Fukuoka et al., 2004; Reisman et al., 2003). SMARCA4 mutations have also been noted in 35% of NSCLC cell lines, as well as in medulloblastoma, Burkitt lymphoma, melanoma, HCC, ccRCC, HNSCC, RT, pancreatic, breast, and prostate cancer (Cancer Genome Atlas Research Network, 2013; Endo et al., 2013; Love et al., 2012; Medina et al., 2008; Oike et al., 2013; Shain and Pollack, 2013). The majority of mutations and deletions are homozygous, though loss of only one allele may be sufficient to drive tumorigenesis. For instance, 10% of mice heterozygous for SMARCA4 develop mammary tumors without spontaneous loss of the second allele (Bultman et al., 2000; Bultman et al., 2008). Functional studies have so far identified dual roles for SMARCA4 in both differentiation and cell adhesion/migration. In embryonic stem cells (ESCs), inactivation of SMARCA4 leads to defective self-renewal and promotes differentiation, while overexpression enhances the epigenetic reprogramming of fibroblasts into induced pluripotent stem (iPS) cells, possibly through increased OCT4 binding to target genes (Singhal et al., 2010). In addition, SMARCA4 has been shown to promote osteoblast differentiation (Flowers et al., 2009). Alternatively, SMARCA4 overexpression in cervical cancer cell lines leads to increased ROCK1 and stress fiber formation, which is reversible upon SMARCA4 knockdown (Asp et al., 2002). Through this latter mechanism, SMARCA4 may serve to modulate cell migration and reduce the potential for invasion.
SMARCA2, the other ATPase subunit of the SWI/SNF complex, is also a bona fide tumor suppressor with frequent loss of expression via epigenetic silencing and more recently, mutational inactivation (Oike et al., 2013). Mouse models have shown that SMARCA2 deficiency results in proliferative abnormalities, including increased overall weight and tissue-specific increased growth (Reyes et al., 1998). These mice exhibit increased cell proliferation in the prostate as well as androgen independence (Shen et al., 2008). In a similar lung model, heterozygous or homozygous loss of SMARCA2 led to increased tumor formation (Glaros et al., 2007). Other studies have proposed another mechanism for enhanced tumorigenicity following SMARCA2 or SMARCA4 loss, specifically through the induction of an epithelial-mesenchymal transition (EMT) phenotype via the transcription factor ZEB1 (Matsubara et al., 2013; Sanchez-Tillo et al., 2010). Clinically, SMARCA2 inactivation may predict poor prognosis, as loss of expression in both NSCLC and HCC correlates with worse OS (Endo et al., 2013; Fukuoka et al., 2004; Reisman et al., 2003). Recently, we identified somatic nonsynonymous mutations in SMARCA2 (5%) following whole-exome and whole-genome sequencing of 60 patients with ACC (Ho et al., 2013). All of these mutations were located in a region encoding the Helicase C domain, which is involved in regulating gene transcription. Other SMARCA2 mutations have also been reported in another cohort of patients with melanoma (Nikolaev et al., 2012). Despite epigenetic silencing being the dominant mechanism for SMARCA2 inactivation, specific mutations may also prove important in the pathogenesis of certain cancers.
PBRM1
Mutations in chromatin state regulators are very common in ccRCC, with PBRM1/BAF180 mutations noted in 29%–41% of all ccRCCs (Hakimi et al., 2013a; Varela et al., 2011). In fact, the only gene altered more frequently in this malignancy is VHL, a well-characterized driver of these cancers. In addition, lower frequency mutation of PBRM1 has also been described in many other neoplasms, including DLBCL, HNSCC, chronic lymphoid leukemia (CLL), gastric, pancreatic, and breast cancer (Shain and Pollack, 2013; Xia et al., 2008). Many of these mutations are truncating—frameshift and nonsense—though a sizable number are also missense, evenly distributed across the protein (Hakimi et al., 2013a). It is most likely that these mutations are inactivating, as loss of PBRM1 expression and gene deletion are noted in several cancers and cell lines (Wang et al., 2012). Further, functional studies have shown that re-expression of PBRM1 in PBRM1-deficient cells induces cell growth arrest in G1 phase, specifically through binding of the CDKN1A promoter, leading to induction of p21 (Xia et al., 2008). PBRM1 contains six tandem bromodomains that bind acetylated histone, two BAH domains that mediate protein-protein interactions, and a HMG domain to bind nucleosomal DNA, all of which provide functional specificity to SWI/SNF remodelers (Wilson and Roberts, 2011).
Exactly how these mutations affect epigenetic reprogramming has yet to be determined, though they are likely pleiotropic, diverse, and dependent on location and type of mutation. It is clear that PBRM1 inactivation does have clinical consequences, at least in ccRCC. For example, PBRM1 mutations have been shown to correlate to worse clinical outcomes (Pawlowski et al., 2013). Also, patients with PBRM1 inactivation are more likely to present with stage III or IV disease, increased tumor size, low differentiation grade, increased perinephric or lymphatic invasion, and are less likely to have an “incidentaloma” at diagnosis (da Costa et al., 2013; Hakimi et al., 2013a; Pawlowski et al., 2013). In addition, smaller tumors (<4 cm) with PBRM1 mutations are more likely to exhibit stage II pathologic features. Most interestingly, PBRM1 and BAP1 mutations are mutually exclusive in ccRCC, and in a side-by-side comparison, PBRM1 mutation correlates with improved OS over BAP1 alterations (Kapur et al., 2013; Pena-Llopis et al., 2013).
BRD7
BRD7 is a PBAF-specific chromatin remodeler that is frequently deleted in breast cancer. Recently, low frequency mutations in this gene have been identified in several cancers (Cerami et al., 2012; Forbes et al., 2011). For some time, BRD7 was believed to be an important SWI/SNF tumor suppressor that functions mainly as a cofactor for p53 to activate oncogene-induced senescence (OIS) (Burrows et al., 2010). In fact, BRD7 was first identified in a loss-of-function screen for genes required for p53-dependent OIS. Drost and colleagues showed that p53 required BRD7 for OIS—and vice versa—and that p53 directly interacts with the N-terminus of BRD7, upstream of its bromodomain. Furthermore, BRD7 was required for induction of several p53 target genes, including CDKN1A and MDM2, and was capable of direct binding to their associated promoters (Drost et al., 2010). This mechanism likely explains the reported ability of BRD7 to inhibit cell cycle progression from G1 to S phase, though direct binding at the E2F3 promoter may also facilitate this (Peng et al., 2006).
Though this mechanism is likely important in certain cancers, the diversity of BRD7 phenotypes may be dependent on the unique molecular architecture found within different tissues. For example, a recent study focusing on epithelial ovarian carcinoma showed that BRD7 can act as a tumor suppressor independent of p53 activity, possibly by sequestering β-catenin in the cytoplasm. These data were further supported by a β-catenin responsive TCF-reporter assay in vitro and the presence of decreased BRD7 expression in high-grade epithelial ovarian serous carcinoma clinical specimens (Bae et al., 2013). Regardless of the mechanism, overexpression of BRD7 has powerful tumor suppressive effects in cancer cell lines, resulting in decreased cell viability and reduced invasion/migration, independent of p53 status. Lastly, BRD7 may also influence covalent histone modifications themselves, with knockdown resulting in decreased acetylation at H3K9 and direct binding observed in vitro at histone residue H3K14 (Bae et al., 2013; Peng et al., 2006). Future studies are necessary to determine which mutations have functional consequences and whether any associated therapeutic vulnerabilities can be exploited.
HISTONES
An exciting development in the past few years has been the identification of recurrent mutations in genes encoding the histones themselves. Histone mutations in cancer were first discovered following massive exomic sequencing efforts in colon cancer (HIST1H1B, 4%) and NHL (HIST1H1C, 7%), though frequencies were low and no recurrent mutations were identified (Morin et al., 2011; Sjoblom et al., 2006). However, a recent pair of pediatric glioma studies revealed highly recurrent heterozygous mutations in HIST1H3B and H3F3A, which encode histones H3.1 and H3.3, respectively. Collectively, Wu et al. and Schwartzentruber et al. reported common H3F3A mutations in diffuse intrinsic pontine glioma (DIPG; 78%) and pediatric GBM (22%–31%), with HIST1H3B mutations almost exclusive to DIPG (18%). Most striking is the location of these mutations at amino acid residues K27M and G34 (G/R, G/V), which are at or near critical sites for post-translational modification via methylation and acetylation on the histone tail (e.g. H3K27, H3K36) (Schwartzentruber et al., 2012; Wu et al., 2012). This observation, along with the mutually exclusive and monoallelic nature of the K27 and G34 mutations, led to subsequent investigations into the clinical and biologic consequences of these potentially gain-of-function mutations.
Currently, K27M and G34R/V mutations have been found almost exclusively in pediatric HGGs, including CNS-PNET (11%, G34R), though sporadic mutations at these residues have been recently noted in osteosarcoma (G34W) and MDS (K27N) (Attieh et al., 2013; Gessi et al., 2013; Joseph et al., 2013). In fact, the remarkable prevalence of these mutations in pediatric HGGs has enabled a thorough examination into the clinical significance of these events. For example, despite the mutually exclusive nature of K27M and G34 mutations, which implies a common pathway or biological process, there are important differences. First, several studies have noted that the K27M mutation predicts both younger age and worse OS as compared to G34 and wild-type H3.3 (Chan et al., 2013a; Khuong-Quang et al., 2012; Sturm et al., 2012). In one study, G34 mutations were actually associated with improved OS when compared to wild-type H3.3 (Sturm et al., 2012). Second, H3F3A mutations are frequently exclusive with IDH1 mutations and commonly co-occur with ATRX/DAXX, PDGFRA, and TP53 alterations, especially in G34 mutant tumors (Schwartzentruber et al., 2012; Sturm et al., 2012). Overlap between alterations in H3F3A and ATRX/DAXX is particularly interesting, since both have been linked to increased CNVs and alternative lengthening of telomeres (ALT) in tumor specimens (Yuen and Knoepfler, 2013). Third, H3F3A mutations predict tumor anatomic location—K27M and G34 mutant GBMs occur in midline and hemispheric areas of the brain, respectively (Chan et al., 2013a; Schwartzentruber et al., 2012). Likewise, a groundbreaking study by Sturm and colleagues revealed distinct epigenetic subtypes of GBM, with K27 and G34 mutant tumor samples exhibiting independent gene expression and methylation profiles (Bjerke et al., 2013; Schwartzentruber et al., 2012; Sturm et al., 2012). Interestingly, these profiles may also explain the anatomic preferences; the G34 mutant transcriptomic signature resembles embryonic regions of the neocortex and striatum, whereas the K27M signature is more similar to embryonic regions of the striatum and thalamus. Lastly, these epigenetic signatures harbor important differences in marker genes, with DNA hypermethylation and associated silencing at FOXG1 (K27M), MICA (K27M), or OLIG1/2 (G34) and elevated expression of PDGFRA (K27M) (Bender et al., 2013; Sturm et al., 2012). Collectively, these changes in the methylome and transcriptome define the different H3.3 mutant GBM subtypes, correlate to anatomic tumor locations, and indicate potentially different cell origins or initiation events in tumorigenesis. Further, they provide mechanistic insight into H3F3A-induced oncogenesis.
Global DNA hypomethylation and decreases in genome-wide histone methylation suggest that mutant H3.3 functions in a dominant-negative manner (Bender et al., 2013; Chan et al., 2013a; Lewis et al., 2013; Sturm et al., 2012). Several groups have now shown that K27M mutant patient samples and cell lines exhibit globally reduced H3K27 di-/tri-methylation and at many loci, DNA hypomethylation (Bender et al., 2013; Chan et al., 2013a; Lewis et al., 2013). Further, forced overexpression of H3.3 K27M in isogenic GBM cells, 293T cells, and MEFs causes reduced H3K27me2/me3 (but not changes in monomethylation) as well as modest increases in H3K27ac, two features of transcriptionally active chromatin (Bender et al., 2013; Chan et al., 2013b). The major mechanism for loss of histone di-/tri-methylation is K27M-mediated inhibition of the PRC2 complex through direct binding of KMT EZH2 at the catalytic site, as well as binding to PRC2 cofactor SUZ12 (Bender et al., 2013; Chan et al., 2013a; Lewis et al., 2013). In fact, in vitro KMT assays revealed that K27M reduces KMT activity by 40%–70% and EZH2 catalytic activity up to 85%, with no loss of PRC2-associated KDM activity (e.g KDM6A/B) (Bender et al., 2013). Somewhat contradictory to this finding, ChIP-seq has revealed that hundreds of gene loci are also enriched for H3K27me3 and EZH2 in K27M mutant tumors or following overexpression of K27M, and analysis of differential expression shows increased gene transcription at these loci (Bender et al., 2013; Bjerke et al., 2013; Chan et al., 2013a). The mechanism underlying these paradoxical changes in H3K27 methylation and associated transcription changes is currently unknown (Chan et al., 2013b).
In contrast to the K27M mutation, broad changes in DNA methylation and histone marks underlying G34R/V induced tumorigenesis are less clear. Further, though investigators have shown that G34R/V mutations disrupt methylation at nearby amino acid residue K36, our understanding of the mechanisms leading to malignant transformation is limited. Despite this, analysis of differential gene expression between H3.3 G34R/V mutant and wild-type tumors has already identified some interesting markers (e.g. OLIG1/2), as stated above. As such, the defining feature of H3.3 mutant tumors is the aberrant activation and repression of numerous genes that uniquely contribute to oncogenesis. Understanding the constellation of changes unique to H3.3 mutants should allow identification of additional subtypes and driver genes. Most exciting is the recent identification of G34-induced activation of MYCN in pediatric GBM cell line KNS42 (Bjerke et al., 2013). This well-characterized oncogene is potentially druggable through synthetic lethal means, via aurora kinase A (AURKA), or JQ1 inhibition (Huang and Weiss, 2013). Implementing this strategy in patients harboring H3F3A G34 mutations could have immense therapeutic benefit. Similarly, identifying and characterizing other differentially expressed genes in H3F3A mutant pediatric gliomas could present new ways of treating this very difficult disease.
SUMMARY AND PERSPECTIVES
Understanding the role of altered epigenetic states has long been a fundamental goal in cancer research. It is now well-documented that distinct chromatin states are both necessary and sufficient to drive tumor formation, sustain increased cellular growth, and encourage metastatic dissemination (Baylin et al., 2001; Jones and Baylin, 2002). However, the link between classical genetics and cancer epigenomics has only recently been explored. In recent years, a plethora of mutations have been discovered in chromatin modifier genes. Studies now show that these alterations have profound effects on the cancer epigenome, leading to potent oncogenic transcriptional programs.
It has become abundantly clear that mutations in epigenetic modifiers are both incredibly diverse and ubiquitous in cancer. In fact, sequencing data has revealed that mutations exist in genes involved in nearly all aspects of epigenetics, including DNA methylation, covalent histone modification, and chromatin remodeling (Fig. 2). Several neoplasms rely on few, yet powerful, mutations in select genes to drive tumorigenesis through an altered epigenome, at least in part (e.g. IDH1/2 in LGG, MLL2 in FL, SMARCB1 in RT). Remarkably, some cancers have such a wide range and high prevalence of alterations in chromatin modifiers (ACC, ccRCC, TCC) that they may be driven primarily through epigenetic means. Interestingly, mutations in regulators of DNA methylation have a strong cancer-specific prevalence (glioma, leukemia), whereas genetic alterations in histone modifiers (e.g. KDM6A) are widespread across many cancers, though the significance of this is incompletely understood.

Frequently mutated epigenetic regulators in human cancer. Select oncogenes and tumor suppressors implicated in altered cancer epigenomes are shown, along with mutation frequency by tumor histology. *Role in oncogenesis (oncogene vs. tumor suppressor) is either mixed or undetermined
Although the field of epigenomics in cancer is relatively new, the identification of driver mutations in epigenetic regulator genes has already led to new prognostic and therapeutic advances. New subtypes of cancer have been identified, or at least attributed to previously unknown genetic alterations, allowing a more nuanced approach to treatment. Further, several new targeted therapies are currently in development and show great promise at reversing phenotypes caused by activating mutations in certain genes (e.g. IDH1, EZH2). In addition, understanding the epigenetic mechanisms that underlie these alterations has also provided new therapeutic potential, even if the driver mutations cannot be targeted directly (e.g. HDACi, DNMT inhibitors) (Table 1) (Arrowsmith et al., 2012; Foulks et al., 2012; Helin and Dhanak, 2013; New et al., 2012; Plass et al., 2013).
There is no doubt that we are in the midst of an exciting era in both cancer genomics and epigenetics. Investigations focused on the intersection of these two fields have provided remarkable clarity into numerous aspects of cancer biology and novel mechanisms of oncogenesis. Already, great strides have been made to translate these early discoveries into clinical practice, and the characterization of novel mutations in chromatin modifier genes is rapidly accelerating. With the ongoing development of new epigenetic therapies, it is possible that much improved outcomes, at least in certain cancers, may be possible in the near future. Further, an enhanced understanding of the epigenomic alterations associated with these driver gene mutations could lead to the discovery of novel pathways involved in tumorigenesis, which may themselves possess unique vulnerabilities for therapeutic intervention.
Abbreviations
- ACC:
-
adenoid cystic carcinoma
- AML:
-
acute myeloid leukemia
- CGI:
-
CpG island
- HNPCC:
-
hereditary nonpolyposis colon cancer
- HDACs:
-
histone deacetylatases
- MSI:
-
microsatellite instability
- TCC:
-
transitional cell carcinoma
References
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R et al (2009) Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114:144–147
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, Beran M, Gilliland DG, Levine RL, Tefferi A (2011) Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 25:1200–1202
Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T et al (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224:334–343
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11:384–400
Asp P, Wihlborg M, Karlen M, Farrants AK (2002) Expression of BRG1, a human SWI/SNF component, affects the organisation of actin filaments through the RhoA signalling pathway. J Cell Sci 115:2735–2746
Attieh Y, Geng QR, Dinardo CD, Zheng H, Jia Y, Fang ZH, Ganan-Gomez I, Yang H, Wei Y, Kantarjian H et al (2013) Low frequency of H3.3 mutations and upregulated DAXX expression in MDS. Blood 121:4009–4011
Bachman KE, Rountree MR, Baylin SB (2001) Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem 276:32282–32287
Bae DS, Park YA, Lee JW, Kim HS, Lee YY, Kim TJ, Choi C, Choi JJ, Jeon HK, Cho YJ et al (2013) Tumor suppressive effects of bromodomain containing protein 7 (BRD7) in epithelial ovarian carcinoma. Clin Cancer Res
Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A (2007) Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res 67:3545–3550
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Bannister AJ, Kouzarides T (1996) The CBP co-activator is a histone acetyltransferase. Nature 384:641–643
Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG (2001) Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 10:687–692
Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T et al (2013) EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23:677–692
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W et al (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24:660–672
Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C et al (2011) The genomic complexity of primary human prostate cancer. Nature 470:214–220
Bisling KE, Brewin JN, McGovern AP, Horne GA, Rider T, Stewart HJ, Ramsahoye BH, Chevassut TJ (2013) DNMT3A mutations at R882 hotspot are only found in major clones of acute myeloid leukemia. Leuk Lymphoma 1:4
Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M et al (2013) Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 3:512–519
Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A et al (2013) Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500:222–226
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A et al (2009) IDH1 mutations at residue p. R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11
Bodor C, O’Riain C, Wrench D, Matthews J, Iyengar S, Tayyib H, Calaminici M, Clear A, Iqbal S, Quentmeier H et al (2011) EZH2 Y641 mutations in follicular lymphoma. Leukemia 25:726–729
Bodor C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, Marzec J, Araf S, Wang J, Lee AM et al (2013) EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 122:3165–3168
Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM et al (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17:72–79
Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA et al (2013) 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 4:1737–1747
Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM et al (1996) The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet 14:33–41
Bosse T, Ter Haar NT, Seeber LM, Diest PJ, Hes FJ, Vasen HF, Nout RA, Creutzberg CL, Morreau H, Smit VT (2013) Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol 26:1525–1535
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22:5323–5335
Brecqueville M, Cervera N, Gelsi-Boyer V, Murati A, Adelaide J, Chaffanet M, Rey J, Vey N, Mozziconacci MJ, Birnbaum D (2011) Rare mutations in DNMT3A in myeloproliferative neoplasms and myelodysplastic syndromes. Blood Cancer J 1:e18
Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G et al (2000) A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell 6:1287–1295
Bultman SJ, Herschkowitz JI, Godfrey V, Gebuhr TC, Yaniv M, Perou CM, Magnuson T (2008) Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 27:460–468
Burrows AE, Smogorzewska A, Elledge SJ (2010) Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci USA 107:14280–14285
Cairns RA, Mak TW (2013) Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3:730–741
Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499:43–49
Capello D, Gloghini A, Martini M, Spina M, Tirelli U, Bertoni F, Rinaldi A, Morra E, Rambaldi A, Sinigaglia F et al (2011) Mutations of CD79A, CD79B and EZH2 genes in immunodeficiency-related non-Hodgkin lymphomas. Br J Haematol 152:777–780
Capper D, Simon M, Langhans CD, Okun JG, Tonn JC, Weller M, von Deimling A, Hartmann C (2012) 2-Hydroxyglutarate concentration in serum from patients with gliomas does not correlate with IDH1/2 mutation status or tumor size. Int J Cancer 131:766–768
Caramel J, Quignon F, Delattre O (2008) RhoA-dependent regulation of cell migration by the tumor suppressor hSNF5/INI1. Cancer Res 68:6154–6161
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y et al (2012) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44:23–31
Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R et al (2013a) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27:985–990
Chan KM, Han J, Fang D, Gan H, Zhang Z (2013b) A lesson learned from the H3.3K27M mutation found in pediatric glioma: a new approach to the study of the function of histone modifications in vivo? Cell Cycle 12:2546–2552
Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, Wichmann M, Schwarzer A, Preller M, Thol F et al (2013) Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122:2877–2887
Chedin F, Lieber MR, Hsieh CL (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci USA 99:16916–16921
Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, Ward PS, Bradner JE, Thompson C, Lowe SW (2013a) Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev 27:1974–1985
Chen Q, Chen Y, Bian C, Fujiki R, Yu X (2013b) TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493:561–564
Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV (1999) c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22:102–105
Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D’Alessio AC, Hysolli E, Guo Y, Yao H, Megyola CM et al (2013) An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep 5:471–481
Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL (1998) The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92:3025–3034
Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M et al (2007) PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC et al (2010) Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 115:2749–2754
Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, Lee MC, Ko BS, Tang JL, Yao M et al (2011a) TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 118:3803–3810
Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY et al (2011b) The prognostic impact and stability of isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 25:246–253
Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128:1063–1076
Christiaans I, Kenter SB, Brink HC, van Os TA, Baas F, van den Munckhof P, Kidd AM, Hulsebos TJ (2011) Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet 48:93–97
Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, Fidalgo M, Saunders A, Lawrence M, Dietmann S et al (2013) NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495:370–374
Croonquist PA, Van Ness B (2005) The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 24:6269–6280
da Costa WH, Rezende M, Rocha RM, da Cunha IW, Carraro DM, Guimaraes GC, de Cassio Zequi S (2013) PBRM1, a SWI/SNF complex subunit is a prognostic marker in clear cell renal cell carcinoma. BJU Int
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C et al (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463:360–363
Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E (2000) The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol 20:3137–3146
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Datta J, Ghoshal K, Sharma SM, Tajima S, Jacob ST (2003) Biochemical fractionation reveals association of DNA methyltransferase (Dnmt) 3b with Dnmt1 and that of Dnmt 3a with a histone H3 methyltransferase and Hdac1. J Cell Biochem 88:855–864
Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
Delhommeau F, Dupont S, James C, Masse A, le Couedic J, Valle V (2008) TET2 is a novel tumor suppressor gene inactivated in myeloproliferative neoplasms: identification of a pre-JAK2 V617F event. Blood 112:11
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A et al (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360:2289–2301
Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E et al (2013) TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32:645–655
Dimartino JF, Cleary ML (1999) Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol 106:614–626
DiNardo CD, Propert KJ, Loren AW, Paietta E, Sun Z, Levine RL, Straley KS, Yen K, Patel JP, Agresta S et al (2013) Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 121:4917–4924
Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, Fries T, Kronke J, Kuhn MW, Paschka P et al (2012) Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 120:e83–e92
Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R et al (2010) BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol 12:380–389
Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM, Kok K (2010) Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res 70:4287–4291
Duns G, Hofstra RM, Sietzema JG, Hollema H, van Duivenbode I, Kuik A, Giezen C, Jan O, Bergsma JJ, Bijnen H et al (2012) Targeted exome sequencing in clear cell renal cell carcinoma tumors suggests aberrant chromatin regulation as a crucial step in ccRCC development. Hum Mutat 33:1059–1062
Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev 8:869–884
Edmunds JW, Mahadevan LC, Clayton AL (2008) Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 27:406–420
Ehrlich M (2002) DNA methylation in cancer: too much, but also too little. Oncogene 21:5400–5413
Ellis L, Atadja PW, Johnstone RW (2009) Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther 8:1409–1420
El-Osta A (2004) The rise and fall of genomic methylation in cancer. Leukemia 18:233–237
Endo M, Yasui K, Zen Y, Gen Y, Zen K, Tsuji K, Dohi O, Mitsuyoshi H, Tanaka S, Taniwaki M et al (2013) Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int 33:105–117
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A et al (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42:722–726
Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, Trojan J, Vaurs-Barriere C, Bignon YJ, Ramus S et al (2001) DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 10:3001–3007
Ewalt M, Galili NG, Mumtaz M, Churchill M, Rivera S, Borot F, Raza A, Mukherjee S (2011) DNMT3a mutations in high-risk myelodysplastic syndrome parallel those found in acute myeloid leukemia. Blood Cancer J 1:e9
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301:89–92
Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12:1052–1058
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567
Flanagan S, Lee M, Li CC, Suter CM, Buckland ME (2012) Promoter methylation analysis of IDH genes in human gliomas. Front Oncol 2:193
Flowers S, Nagl NG Jr, Beck GR Jr, Moran E (2009) Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation. J Biol Chem 284:10067–10075
Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S et al (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 125:659–669
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A et al (2011) COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 39:D945–D950
Foulks JM, Parnell KM, Nix RN, Chau S, Swierczek K, Saunders M, Wright K, Hendrickson TF, Ho KK, McCullar MV et al (2012) Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen 17:2–17
Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, Tsark WM, Riggs AD, Ku HT, Huang W (2013) MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci USA 110:17892–17897
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F et al (2012) Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 44:760–764
Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J 20:2536–2544
Fuks F, Hurd PJ, Deplus R, Kouzarides T (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31:2305–2312
Fukuoka J, Fujii T, Shih JH, Dracheva T, Meerzaman D, Player A, Hong K, Settnek S, Gupta A, Buetow K et al (2004) Chromatin remodeling factors and BRM/BRG1 expression as prognostic indicators in non-small cell lung cancer. Clin Cancer Res 10:4314–4324
Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M (1983) The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11:6883–6894
Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z (2008) ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA 105:6656–6661
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1
Ge YZ, Pu MT, Gowher H, Wu HP, Ding JP, Jeltsch A, Xu GL (2004) Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J Biol Chem 279:25447–25454
Geisbrecht BV, Gould SJ (1999) The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem 274:30527–30533
Gessi M, Gielen GH, Hammes J, Dorner E, Muhlen AZ, Waha A, Pietsch T (2013) H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: possible diagnostic and therapeutic implications? J Neurooncol 112:67–72
Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A, Mak TW, Bristow RG (2012) IDH mutation status in prostate cancer. Oncogene 31:3826
Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D (2007) The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene 26:7058–7066
Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG et al (2000) Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 26:16–17
Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, Astley H, Gitterman D, Henley T, Howes R et al (2012) Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J Biol Chem 287:42180–42194
Green A, Beer P (2010) Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 362:369–370
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM et al (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207:339–344
Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, Haferlach T, Schnittger S (2011) Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia 25:877–879
Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, Stadler K, Bellos F, Kern W, Haferlach T et al (2013) The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosom Cancer 52:410–422
Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595–606
Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV et al (2011) EZH2 mutational status predicts poor survival in myelofibrosis. Blood 118:5227–5234
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L et al (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43:875–878
Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, Smith TW, Imbalzano AN, Jones SN (2001) Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol 21:3598–3603
Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, Gomez V, Hergovich A, Tirabosco R, Fernanda Amary M et al (2013) Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun 4:2166
Guo JU, Su Y, Zhong C, Ming GL, Song H (2011) Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145:423–434
Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, Liu H, Takeda S, Tickoo SK, Reuter VE et al (2013a) Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol 63:848–854
Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK et al (2013b) Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 19:3259–3267
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hanigan CL, Van Engeland M, De Bruine AP, Wouters KA, Weijenberg MP, Eshleman JR, Herman JG (2008) An inactivating mutation in HDAC2 leads to dysregulation of apoptosis mediated by APAF1. Gastroenterology 135(1654–1664):e1652
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L et al (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307
Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA et al (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39:311–318
Helin K, Dhanak D (2013) Chromatin proteins and modifications as drug targets. Nature 502:480–488
Herman JG, Baylin SB (2003) Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349:2042–2054
Hermann A, Gowher H, Jeltsch A (2004) Biochemistry and biology of mammalian DNA methyltransferases. CMLS 61:2571–2587
Herz HM, Madden LD, Chen Z, Bolduc C, Buff E, Gupta R, Davuluri R, Shilatifard A, Hariharan IK, Bergmann A (2010) The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol Cell Biol 30:2485–2497
Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT et al (2013) The mutational landscape of adenoid cystic carcinoma. Nat Genet 45:791–798
Holliday R, Pugh JE (1975) DNA modification mechanisms and gene activity during development. Science 187:226–232
Holz-Schietinger C, Matje DM, Harrison MF, Reich NO (2011) Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J Biol Chem 286:41479–41488
Holz-Schietinger C, Matje DM, Reich NO (2012) Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 287:30941–30951
Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC et al (2012) DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood 119:559–568
Hsieh CL (1999) In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol Cell Biol 19:8211–8218
Huang M, Weiss WA (2013) G34, another connection between MYCN and a pediatric tumor. Cancer Discov 3:484–486
Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA et al (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 13:587–597
Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P (2007) Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet 80:805–810
Hussein K, Abdel-Wahab O, Lasho TL, Van Dyke DL, Levine RL, Hanson CA, Pardanani A, Tefferi A (2010) Cytogenetic correlates of TET2 mutations in 199 patients with myeloproliferative neoplasms. Am J Hematol 85:81–83
Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N (2002) Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J Biol Chem 277:41674–41685
Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, Wang X, Biegel JA, Pomeroy SL, Mesirov JP et al (2005) Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci USA 102:17745–17750
Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E (2007) Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol 27:1889–1903
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466:1129–1133
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303
Iyer NG, Ozdag H, Caldas C (2004) p300/CBP and cancer. Oncogene 23:4225–4231
Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP et al (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 45:1386–1391
Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M et al (2010) Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 16:1429–1433
Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O’Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP (2009) Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 113:6403–6410
Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449:248–251
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128:683–692
Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B et al (2010) Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330:228–231
Joseph CG, Hwang H, Jiao Y, Wood LD, Kinde I, Wu J, Mandahl N, Luo J, Hruban RH, Diaz LA Jr et al (2013) Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosom Cancer 53:15–24
Jurkowska RZ, Rajavelu A, Anspach N, Urbanke C, Jankevicius G, Ragozin S, Nellen W, Jeltsch A (2011) Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J Biol Chem 286:24200–24207
Kampranis SC, Tsichlis PN (2009) Histone demethylases and cancer. Adv Cancer Res 102:103–169
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J et al (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466:869–873
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 57:808–811
Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429:900–903
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH (2009) Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125:353–355
Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK, Xie XJ, Brugarolas J (2013) Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol 14:159–167
Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B et al (2013) Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 27:1301–1309
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447
Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP (2008) SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol 28:3457–3464
Kim JG, Takeshima H, Niwa T, Rehnberg E, Shigematsu Y, Yoda Y, Yamashita S, Kushima R, Maekita T, Ichinose M et al (2013) Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers. Cancer Lett 330:33–40
Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ (2004) Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev 18:1592–1605
Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF et al (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100:11606–11611
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J et al (2012) A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8:890–896
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R et al (2010) Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468:839–843
Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H et al (2013) Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497:122–126
Konstandin N, Bultmann S, Szwagierczak A, Dufour A, Ksienzyk B, Schneider F, Herold T, Mulaw M, Kakadia PM, Schneider S et al (2011) Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia 25:1649–1652
Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguie F, Quesnel B, Beyne-Rauzy O, Solary E et al (2009) TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 114:3285–3291
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Kreiger PA, Judkins AR, Russo PA, Biegel JA, Lestini BJ, Assanasen C, Pawel BR (2009) Loss of INI1 expression defines a unique subset of pediatric undifferentiated soft tissue sarcomas. Mod Pathol 22:142–150
Krivtsov AV, Armstrong SA (2007) MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7:823–833
Kung AL, Rebel VI, Bronson RT, Ch’ng LE, Sieff CA, Livingston DM, Yao TP (2000) Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev 14:272–277
Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA et al (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44:609–620
Laffaire J, Everhard S, Idbaih A, Criniere E, Marie Y, de Reynies A, Schiappa R, Mokhtari K, Hoang-Xuan K, Sanson M et al (2011) Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neuro-oncology 13:84–98
Lai JL, Jouet JP, Bauters F, Deminatti M (1985) Chronic myelogenous leukemia with translocation (8;22): report of a new case. Cancer Genet Cytogenet 17:365–366
Larkin J, Goh XY, Vetter M, Pickering L, Swanton C (2012) Epigenetic regulation in RCC: opportunities for therapeutic intervention? Nat Rev Urol 9:147–155
Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318:447–450
Lee JJ, Geli J, Larsson C, Wallin G, Karimi M, Zedenius J, Hoog A, Foukakis T (2008) Gene-specific promoter hypermethylation without global hypomethylation in follicular thyroid cancer. Int J Oncol 33:861–869
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340:857–861
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J et al (2010) DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363:2424–2433
Li YQ, Zhou PZ, Zheng XD, Walsh CP, Xu GL (2007) Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res 35:390–400
Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ 3rd, Qiao Q, Neubert TA, Xu RM, Gozani O et al (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 284:34283–34295
Li XS, Trojer P, Matsumura T, Treisman JE, Tanese N (2010) Mammalian SWI/SNF–a subunit BAF250/ARID1 is an E3 ubiquitin ligase that targets histone H2B. Mol Cell Biol 30:1673–1688
Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (2013a) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153:590–600
Li WD, Li QR, Xu SN, Wei FJ, Ye ZJ, Cheng JK, Chen JP (2013b) Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood 121:1478–1479
Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q et al (2012) Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150:1135–1146
Lin J, Yao DM, Qian J, Chen Q, Qian W, Li Y, Yang J, Wang CZ, Chai HY, Qian Z et al (2011) Recurrent DNMT3A R882 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS ONE 6:e26906
Lindberg J, Klevebring D, Liu W, Neiman M, Xu J, Wiklund P, Wiklund F, Mills IG, Egevad L, Gronberg H (2013a) Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur Urol 63:347–353
Lindberg J, Mills IG, Klevebring D, Liu W, Neiman M, Xu J, Wikstrom P, Wiklund P, Wiklund F, Egevad L et al (2013b) The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol 63:702–708
Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL et al (2012) Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA 109:3879–3884
Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR (2003) TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17:637–641
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG Jr (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339:1621–1625
Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, Richards KL, Dunphy CH, Choi WW, Srivastava G et al (2012) The genetic landscape of mutations in Burkitt lymphoma. Nat Genet 44:1321–1325
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478
Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, Intlekofer AM, Chen C, Ye J, Hameed M et al (2013) Induction of sarcomas by mutant IDH2. Genes Dev 27:1986–1998
Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA et al (2008) Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA 105:20380–20385
Majer CR, Jin L, Scott MP, Knutson SK, Kuntz KW, Keilhack H, Smith JJ, Moyer MP, Richon VM, Copeland RA et al (2012) A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett 586:3448–3451
Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, Saunthararajah Y, Guinta K, Keddache MA, Putnam P et al (2010) Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia 24:1799–1804
Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, Edgren H, Kallioniemi O, Aleynikova O, Przybytkowski E et al (2012) An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene 31:2090–2100
Manceau G, Letouze E, Guichard C, Didelot A, Cazes A, Corte H, Fabre E, Pallier K, Imbeaud S, Le Pimpec-Barthes F et al (2013) Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int J Cancer 132:2217–2221
Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ, Chang DK et al (2012) Sleeping beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci USA 109:5934–5941
Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Dohner K, Armstrong SA (2012) Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia 26:1881–1883
Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM et al (2008) The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 111:3145–3154
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, Radmacher MD, Kohlschmidt J, Nicolet D, Whitman SP et al (2012) Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 30:742–750
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Margueron R, Reinberg D (2010) Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11:285–296
Margueron R, Reinberg D (2011) The polycomb complex PRC2 and its mark in life. Nature 469:343–349
Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM et al (2011) The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117:211–220
Matsubara D, Kishaba Y, Ishikawa S, Sakatani T, Oguni S, Tamura T, Hoshino H, Sugiyama Y, Endo S, Murakami Y et al (2013) Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci 104:266–273
McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE et al (2012a) Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA 109:2989–2994
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E et al (2012b) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112
Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M (2008) Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 29:617–622
Minor EA, Court BL, Young JI, Wang G (2013) Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem 288:13669–13674
Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, Hayden MG, Cherry AM, Gokden M, Edwards MS et al (2010) Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol 120:745–753
Morales Torres C, Laugesen A, Helin K (2013) Utx is required for proper induction of ectoderm and mesoderm during differentiation of embryonic stem cells. PLoS ONE 8:e60020
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X et al (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20:11–24
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F et al (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42:181–185
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M et al (2011) Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476:298–303
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J et al (2011) CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471:235–239
Nagl NG Jr, Wang X, Patsialou A, Van Scoy M, Moran E (2007) Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J 26:752–763
Nakajima T, Uchida C, Anderson SF, Parvin JD, Montminy M (1997) Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes Dev 11:738–747
Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ (1998) Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol 15:1674–1684
Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A et al (2013) Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 121:4749–4752
New M, Olzscha H, La Thangue NB (2012) HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol 6:637–656
Newbold RF, Mokbel K (2010) Evidence for a tumour suppressor function of SETD2 in human breast cancer: a new hypothesis. Anticancer Res 30:3309–3311
Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, Dombret H, Dreyfus F, Quesnel B, Geffroy S et al (2010) Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood 116:1132–1135
Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, Wang W (2000) A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol Cell Biol 20:8879–8888
Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V et al (2012) Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 44:133–139
Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T et al (2010) Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42:665–667
Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M, Rini B, Yan Q, Yang H (2012) The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31:776–786
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T (2011) Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30:2135–2146
Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T (2013) Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol 43:849–855
Oka M, Meacham AM, Hamazaki T, Rodic N, Chang LJ, Terada N (2005) De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2’-deoxycytidine. Oncogene 24:3091–3099
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LM, Mohd-Sarip A, Vries RG, Hoeben RC, Verrijzer CP (2004) P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem 279:3807–3816
Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A et al (2013) Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia
Pais A, Amare Kadam P, Raje G, Sawant M, Kabre S, Jain H, Advani S, Banavali S (2005) Identification of various MLL gene aberrations that lead to MLL gene mutation in patients with acute lymphoblastic leukemia (ALL) and infants with acute leukemia. Leuk Res 29:517–526
Panagopoulos I, Fioretos T, Isaksson M, Samuelsson U, Billstrom R, Strombeck B, Mitelman F, Johansson B (2001) Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum Mol Genet 10:395–404
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C et al (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, Spath D, Kayser S, Zucknick M, Gotze K et al (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28:3636–3643
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J et al (2011a) Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471:189–195
Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O et al (2011b) Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 43:830–837
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O et al (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366:1079–1089
Pawlowski R, Muhl SM, Sulser T, Krek W, Moch H, Schraml P (2013) Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int J Cancer 132:E11–E17
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T et al (2012) Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44:1104–1110
Pena-Llopis S, Christie A, Xie XJ, Brugarolas J (2013) Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res 73:4173–4179
Peng C, Zhou J, Liu HY, Zhou M, Wang LL, Zhang QH, Yang YX, Xiong W, Shen SR, Li XL et al (2006) The transcriptional regulation role of BRD7 by binding to acetylated histone through bromodomain. J Cell Biochem 97:882–892
Perez C, Martinez-Calle N, Martin-Subero JI, Segura V, Delabesse E, Fernandez-Mercado M, Garate L, Alvarez S, Rifon J, Varea S et al (2012) TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS ONE 7:e31605
Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ et al (1995) Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376:348–351
Pirozzi CJ, Reitman ZJ, Yan H (2013) Releasing the block: setting differentiation free with mutant IDH inhibitors. Cancer Cell 23:570–572
Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P (2013) Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 14:765–780
Plimack ER, Kantarjian HM, Issa JP (2007) Decitabine and its role in the treatment of hematopoietic malignancies. Leuk Lymphoma 48:1472–1481
Poage GM, Houseman EA, Christensen BC, Butler RA, Avissar-Whiting M, McClean MD, Waterboer T, Pawlita M, Marsit CJ, Kelsey KT (2011) Global hypomethylation identifies loci targeted for hypermethylation in head and neck cancer. Clin Cancer Res 17:3579–3589
Poon SL, Pang ST, McPherson JR, Yu W, Huang KK, Guan P, Weng WH, Siew EY, Liu Y, Heng HL et al (2013) Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med 5:197ra101
Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Masse A, Le Couedic JP, Pendino F, Carbonne B et al (2011) Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 118:2551–2555
Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL et al (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488:106–110
Purdy MM, Holz-Schietinger C, Reich NO (2010) Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch Biochem Biophys 498:13–22
Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH et al (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20:25–38
Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD et al (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593–599
Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE (2003) Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 63:560–566
Reisman D, Glaros S, Thompson EA (2009) The SWI/SNF complex and cancer. Oncogene 28:1653–1668
Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, Cayuela JM, Hayette S, Reman O, Contentin N et al (2012) Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia 26:1247–1254
Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M (1998) Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). EMBO J 17:6979–6991
Riggs AD (1975) X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet 14:9–25
Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH (2000) Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA 97:13796–13800
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48
Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF et al (2006) A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet 38:566–569
Ropero S, Ballestar E, Alaminos M, Arango D, Schwartz S Jr, Esteller M (2008) Transforming pathways unleashed by a HDAC2 mutation in human cancer. Oncogene 27:4008–4012
Ross JS, Wang K, Al-Rohil RN, Nazeer T, Sheehan CE, Otto GA, He J, Palmer G, Yelensky R, Lipson D et al (2013) Advanced urothelial carcinoma: next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod Pathol
Rotili D, Mai A (2011) Targeting histone demethylases: a new avenue for the fight against cancer. Genes Cancer 2:663–679
Rubery ED, Newton AA (1973) DNA methylation in normal and tumour virus-transformed cells in tissue culture. I. The level of DNA methylation in BHK21 cells and in BHK21 cells transformed by polyoma virus (PyY cells). Biochim Biophys Acta 324:24–36
Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, Iafrate AJ, Bernstein BE, Sohani AR, Le LP (2011) EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS ONE 6:e28585
Saha A, Wittmeyer J, Cairns BR (2006) Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 7:437–447
Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A (2010) ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 29:3490–3500
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J et al (2012) D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev 26:2038–2049
Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr, Papadopoulos N et al (2013) Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45:12–17
Schaub FX, Looser R, Li S, Hao-Shen H, Lehmann T, Tichelli A, Skoda RC (2010) Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 115:2003–2007
Schmidt CK, Jackson SP (2013) On your mark, get SET(D2), go! H3K36me3 primes DNA mismatch repair. Cell 153:513–515
Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T (2010) IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 116:5486–5496
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231
Senyuk V, Premanand K, Xu P, Qian Z, Nucifora G (2011) The oncoprotein EVI1 and the DNA methyltransferase Dnmt3 co-operate in binding and de novo methylation of target DNA. PLoS ONE 6:e20793
Shain AH, Pollack JR (2013) The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8:e55119
Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, Maitra A, Pollack JR (2012) Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci USA 109:E252–E259
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31:27–36
Shen JC, Rideout WM 3rd, Jones PA (1992) High frequency mutagenesis by a DNA methyltransferase. Cell 71:1073–1080
Shen H, Powers N, Saini N, Comstock CE, Sharma A, Weaver K, Revelo MP, Gerald W, Williams E, Jessen WJ et al (2008) The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res 68:10154–10162
Shi Y, Whetstine JR (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25:1–14
Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, Yang X, Li Y, Han X, Zhang Y et al (2011) Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA 108:7541–7546
Shiama N (1997) The p300/CBP family: integrating signals with transcription factors and chromatin. Trends Cell Biol 7:230–236
Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N (2011) Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol 178:1395–1402
Shih AH, Abdel-Wahab O, Patel JP, Levine RL (2012) The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12:599–612
Shpargel KB, Sengoku T, Yokoyama S, Magnuson T (2012) UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet 8:e1002964
Sievert AJ, Jackson EM, Gai X, Hakonarson H, Judkins AR, Resnick AC, Sutton LN, Storm PB, Shaikh TH, Biegel JA (2009) Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 19:449–458
Singhal N, Graumann J, Wu G, Arauzo-Bravo MJ, Han DW, Greber B, Gentile L, Mann M, Scholer HR (2010) Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell 141:943–955
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N et al (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314:268–274
Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, Przychodzen B, Mian SA, Nasser EE, Shooter C et al (2010) Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 116:3923–3932
Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA (2010) Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA 107:20980–20985
Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M, Manova-Todorova K, Teruya-Feldstein J, Avigan DE et al (2013) The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 13:87–101
Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, Schauer S, Dohner H, Dohner K (2011) DNMT3A mutations in myeloproliferative neoplasms. Leukemia 25:1217–1219
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A et al (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333:1157–1160
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458:719–724
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437
Suetake I, Mishima Y, Kimura H, Lee YH, Goto Y, Takeshima H, Ikegami T, Tajima S (2011) Characterization of DNA-binding activity in the N-terminal domain of the DNA methyltransferase Dnmt3a. Biochem J 437:141–148
Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H (2007) De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 204:715–722
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L et al (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14:1025–1040
Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Finke CM, Mullally A, Li CY, Pardanani A et al (2009a) Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 23:900–904
Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG, Levine RL (2009b) Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 23:1343–1345
Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A et al (2009c) TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 23:905–911
Thieme S, Gyarfas T, Richter C, Ozhan G, Fu J, Alexopoulou D, Muders MH, Michalk I, Jakob C, Dahl A et al (2013) The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 121:2462–2473
Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D, McCabe NR, Kobayashi H, Ziemin-van der Poel S, Kaneko Y, Morgan R, Sandberg AA et al (1993) Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med 329:909–914
Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, Yun H, Gohring G, Schlegelberger B, Hoelzer D et al (2011a) Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29:2889–2896
Thol F, Winschel C, Ludeking A, Yun H, Friesen I, Damm F, Wagner K, Krauter J, Heuser M, Ganser A (2011b) Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica 96:1870–1873
Timp W, Feinberg AP (2013) Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13:497–510
Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, Sugimoto Y, Szpurka H, Makishima H, O’Keefe CL et al (2013) Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 28:78–87
Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, Grosschedl R (2010) Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription-independent poising of chromatin. Immunity 32:714–725
Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, Gore L, Feusner JH (2009) Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52:328–334
Trotter KW, Archer TK (2004) Reconstitution of glucocorticoid receptor-dependent transcription in vivo. Mol Cell Biol 24:3347–3358
Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV (2005) Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci USA 102:12129–12134
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483
Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, Viale A, Riggins GJ, Chan TA (2013) Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget 4:1729–1736
Vakoc CR, Wen YY, Gibbs RA, Johnstone CN, Rustgi AK, Blobel GA (2009) Low frequency of MLL3 mutations in colorectal carcinoma. Cancer Genet Cytogenet 189:140–141
Valinluck V, Sowers LC (2007) Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 67:946–950
van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O’Meara S, Teague J et al (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41:521–523
Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP et al (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629
Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J et al (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469:539–542
Varier RA, Timmers HT (2011) Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta 1815:75–89
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD et al (1998) Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA 95:8698–8702
Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K et al (2013) Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 49:645–656
Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Viguie F, Aboura A, Bouscary D, Ramond S, Delmer A, Tachdjian G, Marie JP, Casadevall N (2005) Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia 19:1411–1415
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Wakita S, Yamaguchi H, Omori I, Terada K, Ueda T, Manabe E, Kurosawa S, Iida S, Ibaraki T, Sato Y et al (2013) Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia 27:1044–1052
Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O’Laughlin M et al (2011) Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25:1153–1158
Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR (1996) Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev 10:2117–2130
Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ et al (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40:897–903
Wang P, Lin C, Smith ER, Guo H, Sanderson BW, Wu M, Gogol M, Alexander T, Seidel C, Wiedemann LM et al (2009a) Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol 29:6074–6085
Wang X, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PT, Roberts CW (2009b) Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res 69:8094–8101
Wang JK, Tsai MC, Poulin G, Adler AS, Chen S, Liu H, Shi Y, Chang HY (2010) The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev 24:327–332
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY et al (2011) Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet 43:1219–1223
Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, Sang F, Sonoda K, Sugawara M, Saiura A et al (2012) Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res 22:208–219
Wang X, Haswell JR, Roberts CW (2013) Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer-mechanisms and potential therapeutic insights. Clin Cancer Res 20:21–27
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153
Watanabe Y, Castoro RJ, Kim HS, North B, Oikawa R, Hiraishi T, Ahmed SS, Chung W, Cho MY, Toyota M et al (2011) Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PLoS ONE 6:e23320
Webster KE, O’Bryan MK, Fletcher S, Crewther PE, Aapola U, Craig J, Harrison DK, Aung H, Phutikanit N, Lyle R et al (2005) Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc Natl Acad Sci USA 102:4068–4073
Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C, Haferlach T et al (2012) Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 26:934–942
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE et al (2010) ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363:1532–1543
Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, Weller M, Wick W (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126:443–451
Wigle TJ, Knutson SK, Jin L, Kuntz KW, Pollock RM, Richon VM, Copeland RA, Scott MP (2011) The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Lett 585:3011–3014
Wilson BG, Roberts CW (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11:481–492
Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW (2010) Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18:316–328
Winkler BS, DeSantis N, Solomon F (1986) Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Exp Eye Res 43:829–847
Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: what are the cancer relevant targets? Cancer Lett 277:8–21
Wu JN, Roberts CW (2013) ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 3:35–43
Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y (2011) Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev 25:679–684
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253
Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, Memeo L, Hibshoosh H, Parsons R (2008) BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res 68:1667–1674
Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J (2004) Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem 279:33946–33957
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, Ueno T, Soda M, Hamada T, Haruta H et al (2010) Array-based genomic resequencing of human leukemia. Oncogene 29:3723–3731
Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, Kantarjian HM, Estecio MR, Jelinek J, Godley LA et al (2012) Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 7:201–207
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, Shi JY, Zhu YM, Tang L, Zhang XW et al (2011) Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43:309–315
Yan G, Eller MS, Elm C, Larocca CA, Ryu B, Panova IP, Dancy BM, Bowers EM, Meyers D, Lareau L et al (2013) Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J Invest Dermatol 133:2444–2452
Yang YA, Yu J (2013) EZH2, an epigenetic driver of prostate cancer. Protein Cell 4:331–341
Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M et al (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117:2451–2459
Yildirim O, Li R, Hung JH, Chen PB, Dong X, Ee LS, Weng Z, Rando OJ, Fazzio TG (2011) Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147:1498–1510
Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, Zhang D, Huang H, Gao J, Li Z et al (2013) Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc 135:10396–10403
Yokoyama A, Cleary ML (2008) Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14:36–46
Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML (2005) The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123:207–218
You JS, Jones PA (2012) Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 22:9–20
Yuen BT, Knoepfler PS (2013) Histone h3.3 mutations: a variant path to cancer. Cancer Cell 24:567–574
Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A et al (2012) Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet 44:570–574
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M et al (2012) The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481:157–163
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324:261–265
Zhao H, Wang J, Han Y, Huang Z, Ying J, Bi X, Zhao J, Fang Y, Zhou H, Zhou J et al (2011) ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget 2:886–891
Zou P, Xu H, Chen P, Yan Q, Zhao L, Zhao P, Gu A (2013) IDH1/IDH2 mutations define the prognosis and molecular profiles of patients with gliomas: a meta-analysis. PLoS ONE 8:e68782
Acknowledgements
We apologize to the many authors whose work we could not cite because of space constraints. We would like to thank Deepa Ramaswami for help in reviewing the manuscript. D.M.R. was supported by the HHMI Medical Research Fellows Program and NIH MSTP grant T32GM007739. L.A.W. was supported by The Canadian Institutes of Health Research PDF. T.A.C. is a Department of Defense Era of Hope Scholar and supported in part by the Frederick Adler Fund.
Compliance With Ethics Guidelines
Authors David Roy, Logan Walsh, and Timothy Chan declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Roy, D.M., Walsh, L.A. & Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 5, 265–296 (2014). https://doi.org/10.1007/s13238-014-0031-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0031-6