
Abstract
In the spring of 2009 a new triple-reassortant of influenza A (H1N1) virus appeared in Mexico and rapidly spread around the world, becoming a pandemic that primarily infected children and uncommonly older adults. Accompanying the pandemic were associated neurologic and muscular syndromes that affected primarily children and included febrile seizures, encephalopathy/encephalitis with or without seizures, delirium, focal neurologic syndromes, Guillain-Barré syndrome, myositis, and myocarditis. Neither the frequency nor the severity of these syndromes appears different from those recognized during periods of infections of previous influenza A viruses. I review the clinical, laboratory, neuroimaging, and pathologic characteristics of the associated syndromes appearing in the first wave of the pandemic, compare them to similar cases occurring in previous years, and explore several theories of pathogenesis.
Similar content being viewed by others


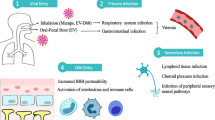
Introduction
In April of 2009 a novel influenza A (H1N1) virus was discovered to cause an outbreak of influenza in Mexico. Soon afterward the virus spread to the United States and then globally to become a pandemic infection. As of March 2010, almost all countries had reported infections, with more than 17,000 deaths reported to the World Health Organization [1•]. In the United States as of mid March 2010, it was clinically estimated that there were 60 million illnesses, 270,000 hospitalizations, and 12,270 deaths caused by the 2009 influenza A virus [2•]. The actual infection rate in children appears to be much higher than the illness estimates. Based on serologic studies, about 1 in 3 children were infected with the virus in the first wave of infection, giving a 10-fold higher incidence rate than that obtained from clinical surveillance [3].
Pathophysiology of Influenza A (H1N1)
Influenza A (H1N1) is a member of the Orthomyxoviridae family and is an enveloped negative-strand RNA virus with six segments. Whereas influenza B and C viruses can infect humans, influenza A is the only virus that historically has caused pandemics [4]. On the viral envelope are two glycoproteins called hemagglutinin (H) and neuraminidase (N), which are important for viral attachment and release from respiratory cells. Combinations of 16 hemagglutinin serotypes and nine neuraminidase serotypes have been identified in wild birds but only three different serotype combinations have widely circulated in humans [4]. Influenza A H1N1 strains have circulated around the world as far back as the famous “Spanish influenza” pandemic of 1918.
The current 2009 influenza A (H1N1) virus has six genes from triple-reassortant North American swine virus lineages and two genes from Eurasian swine virus lineages [1•]. Even though the 2009 H1N1 virus is antigenically distinct from other human and swine influenza A viruses, the highest attack rates of the 2009 strain were reported among children and young adults. The relative sparing of adults older than 60 years appears to be due to the exposure of these adults to antigenically related influenza viruses about six decades earlier resulting in cross-protective antibodies [1•, 3].
Human influenza viruses spread mainly by inhalation of infectious droplets or airborne large droplet nuclei and possibly also by hand to mouth/nose transmission from touching surfaces that contain the influenza virus [4]. Although receptors for human influenza viruses are expressed throughout the respiratory tract from nasal mucosa to alveoli, the highest expression occurs in the upper respiratory tract tracheal and bronchial epithelium, especially the ciliated epithelial cells [1•]. In vivo and in vitro studies also have demonstrated that influenza virus can infect and replicate in nasal mucosa, nasopharynx epithelium, adenoids, tonsils, bronchial epithelium, and pulmonary alveoli. The incubation period for 2009 influenza was 1.4 to 3 days, the same as seasonal influenza [1•]. The peak viral load in the nasopharynx occurs on the day of onset of symptoms and declined gradually over the next 5 to 8 days [1•]. Although experimental animal studies and in vitro studies showed the 2009 H1N1 strain replicated more efficiently than other currently circulating influenza A H1N1 strains, to date complete influenza viral replication has not been demonstrated for influenza A H1N1 virus outside the respiratory tract [5].
Uncomplicated Influenza
Patients with uncomplicated influenza A infections typically experience both respiratory and nonrespiratory symptoms. The respiratory symptoms include nasal congestion, cough, and sore throat that follow viral replication in epithelial respiratory cells [1•, 4]. Nonrespiratory systemic symptoms can include fever, fatigue, vomiting, diarrhea, headache, myalgia, and stiff neck. The severity of these symptoms varies greatly from person to person but the vast majority of influenza cases are acute and self-limited with a case fatality rate of less than 0.5% [1•]. About one half of patients who were hospitalized had no recognized serious coexisting medical conditions, but pulmonary complications appear to be at a higher frequency in pregnant women in the second or third trimester and in patients with pre-existing immunosuppression, asthma, obesity, lung, cardiac disease. or neurologic illness (eg, neurocognitive, neuromuscular, or seizure disorders) [6].
Neurologic Complications Overview
To date, the spectrum and incidence of neurologic complications seen in patients with 2009 influenza appear to be similar to that from other strains of H1N1 influenza except that patients with a complication of Reye’s syndrome have not been published. Published studies of neurologic complications have been limited to case reports or small clinical series and large epidemiologic studies or case series are not yet available. The majority of reported neurologic complications have been in children or young adults and likely represents the much higher viral infection attack rates in these groups. Table 1 lists the major neurologic and muscular illnesses associated with influenza.
Febrile Seizures Associated with Influenza
Febrile seizures are defined as a seizure in children 6 months to 5 years of age associated with fever but without evidence of intracranial infection or other definable cause [7]. In a series of hospital visits for febrile seizures, about 20% are associated with influenza [7, 8]. The majority had a single uncomplicated partial or generalized seizure; however, 20% to 33% had complex seizures (defined as prolonged seizures of >15 min, multiple seizures within 24 h, or prolonged postictal impairment of consciousness for >30 min) [8]. Febrile seizure patients did not have abnormalities on the electroencephalogram (EEG) or neuroimaging and they were discharged as normal. Currently no publications have reported any differences in the frequency or characteristics of febrile seizures associated with the 2009 influenza A.
Encephalopathy with or Without Seizures Associated with Influenza
The encephalopathy with or without seizures appears to be the most common major neurologic association. Typically, patients who develop the encephalopathy do not have pre-existing neurologic problems. The encephalopathy seen with the 2009 influenza A to date had a similar clinical picture as encephalopathies associated with prior strains of influenza A [9•].
Table 2 presents a summary of 32 encephalopathy cases published in detail to date. Most cases have appeared in older children, with the intensity of the encephalopathy ranging from marked lethargy to coma. The majority of cases did not report a viral pneumonia, suggesting that hypoxia or acute respiratory distress syndrome did not account for the encephalopathy. Those with encephalopathy also rarely had myocarditis or myositis, suggesting possible different pathogenesis mechanisms.
Over half the patients developed one or a few seizures that were focal or generalized but some patients experienced status epilepticus. A few children who were reported with a seizure and encephalopathy had only lethargy or mild encephalopathy questioning whether they just had febrile seizures. A few children without encephalopathy developed focal neurologic signs ranging from limb weakness to ataxia.
Patients usually developed signs of influenza with a fever, cough, and myalgias 1 to 4 days before onset of the encephalopathy. The cerebrospinal fluid (CSF) usually contained less than five white blood cells/mm3 with a normal glucose level [10, 11•, 12]. The CSF protein was normal or slightly elevated for the patient’s age. CSF Gram stain and cultures were negative for bacteria. The EEG was normal or nonspecific in 25% but showed diffuse slowing consistent with an encephalopathy in 62% [11•, 12]. Focal spikes were seen in 13%, most often when the patient had status epilepticus. Initial cranial CT scans were normal in 58% and initial MRI scans were normal in 62% but demonstrated focal brain edema in white matter in 31% and bilateral thalamic lesions in 19% [11•, 13]. In several patients, the initial neuroimage was normal but subsequent neuroimaging demonstrated marked thalamic lesions, cerebral edema, or loss of brain substrate [13, 14•]. The encephalopathy duration in most patients was less than 1 week with a good recovery. However, deaths or persistent dementia have developed in patients with a severe encephalopathy, particularly if severe cerebral edema and thalamic lesions are seen on neuroimaging [14•]. Currently there is no evidence that treatment with oseltamivir improves outcome [15].
Reye’s syndrome is a severe encephalopathy, cerebral edema, and acute liver dysfunction that was widely recognized in the 1970s and early 1980s. It is now rare and has not been reported associated with the current pandemic, although children have developed the encephalopathy along with elevated liver transaminases [16].
Delirium Associated with Influenza
Neuropsychiatric behaviors are well associated with influenza A viral infections including the 2009 pandemic [14•, 17]. Children or adolescents present within the first 3 days of influenza with delirium characterized by visual hallucinations, inappropriate laughing or smiling, meaningless words, incoherent speech, and restlessness [14•, 17, 18]. Stupor or coma is not seen. The MRI scan typically is normal or shows a reversible splenium hyperintensity on diffusion-weighted images [18]. The EEG is normal or shows diffuse background slowing or semirhythmic high-voltage slow waves but not seizure spikes [18].
Questions have been raised as to whether administration of oseltamivir to children or the elderly can induce delirious behavior. Based on adverse drug reports submitted to the Japanese Ministry of Health there is still an open question regarding neuropsychiatric events associated with the drug usage [19]. However, Hoffmann-La Roche (Basel, Switzerland), the drug manufacturer, reports that adverse claims regarding neuropsychiatric events are not increased in patients taking oseltamivir [20].
Acute Necrotizing Encephalopathy Associated with Influenza
A 12-year-old girl developed a necrotizing encephalopathy associated with the 2009 influenza A [13]. The girl became comatose over 2 days likely following a seizure. The initial CT scan was normal but the MRI scan the next day demonstrated multifocal symmetric-restricted diffusion abnormalities in the thalami, periventricular white matter of the centrum semiovale, medial temporal lobes, pontine gray matter, and medial cerebellar hemispheres. Brain death occurred on day 3. At autopsy the brain was diffusely enlarged with gross evidence of hemorrhage in the thalami and cerebellum. Histopathology demonstrated coagulation necrosis of neural and glial elements, intraparenchymal hemorrhages, and little inflammation. The meninges were free of inflammation and cortical blood vessels lacked perivascular cuffing with inflammatory cells. No immunohistochemical or polymerase chain reaction evidence of influenza virus was identified in the brain but virus was identified in areas of the lung.
This patient, as well as two other US cases [14•], were similar to cases reported in Japan associated with previous influenza infections [21]. Although acute necrotizing encephalopathy occurs worldwide, it occurs more commonly in Japanese children. The patients often die or survive with mental retardation, permanent dementia or extrapyramidal signs of choreoathetosis, tremors, or parkinsonism.
Acute Myositis and Myopathy Associated with Influenza
Acute myositis associated with influenza infections was originally reported in 1957 with a series of 74 cases [22]. Since then benign acute myositis has been reported in many epidemics of influenza A and B and similar reports have occurred associated with the 2009 influenza A [23, 24]. Most cases develop in school-age boys but myositis does occur in adults [25]. Muscle symptoms usually begin 3 days (range, 1–4) after onset of symptoms of influenza with pain in one or both calves and difficulty in walking. Some patients develop pains in other muscles. The muscles are tender to palpation with occasional focal soft tissue edema but rarely redness of the overlying skin or marked warmth of the muscle. Over 95% of the patients have elevated serum creatine kinase (up to 10-fold) or aspartate aminotransferase levels. Myoglobulinemia or myoglobinuria may also transiently develop. MRI of the involved muscle has demonstrated on T1-weighted images subtle enlargement of the muscle. T2-weighted images and contrast-enhanced images show a patchy high signal of the involved muscle accompanied by fascial edema and subcutaneous edema localized to the tissues overlying the involved muscle [26, 27]. Histopathologic examination of the few reported muscle biopsies shows areas of muscle degeneration, occasional foci of frank necrosis, and minimal inflammatory infiltrates [25, 28]. In mild cases, the muscle fiber changes may be patchy without inflammation suggesting the process was more a myopathy than myositis. Although attempts to isolate influenza virus from the muscle biopsy are often negative, influenza virus has been recovered from biopsies and influenza viral antigens have been demonstrated within degenerating muscle fibers by immunofluorescence [28–30]. Acute muscle pains persist for several days with complete recovery over a week with or without administration of antiviral agents. The differential diagnosis of acute myositis includes general myalgias from uncomplicated influenza, Guillain-Barré syndrome (GBS), and pyomyositis.
Occasionally, the influenza-associated myopathy is severe in adults and children and they develop rhabdomyolysis, a syndrome caused by muscle injury involving the leakage of large quantities of intracellular contents into the plasma [31]. The rhabdomyolysis is characterized by muscles weakness, myalgias, and dark urine. Such patients demonstrate myoglobinemia, myoglobinuria, and markedly elevated serum creatine kinase levels. Complications may include acute renal failure, hyperkalemia, hypocalcemia, hypoalbuminemia, and hyperuricemia [31]. Treatment is usually aggressive fluid administration to reduce the risk of renal failure or alleviate the renal failure. Cases of rhabdomyolysis have been reported associated with the 2009 influenza infections [31, 32]. All had typical clinical pictures and made a full recovery.
Myocarditis Associated with Influenza
Cardiac dysfunction associated with influenza ranges from 0% to 10% depending on the methods used to detect myocardial involvement and occurs through direct effects of the virus on the myocardium or through exacerbation of existing cardiovascular disease [33]. The incidence of myocarditis is highest in hospitalized patients, older adults, and individuals with pre-existing pulmonary or cardiac disease. However, one series of 67 healthy Finnish conscripts hospitalized with influenza A reported that 9% had electrocardiac and echocardiographic evidence of myocarditis [34].
When acute myocarditis presents in a patient with common influenza, the symptoms include chest pains or dyspnea that may also suggest congestive heart failure or pericardial effusion. Cardiac symptoms develop within 1 week of influenza onset. The electrocardiogram demonstrates nonspecific tachycardia (also common in uncomplicated influenza) plus ST-elevation associated with Q waves or occasionally left bundle branch block [35]. Echocardiography usually shows left ventricular dysfunction with wall motion abnormalities and reduced ejection fractions [35, 36]. Cardiac MRI has shown focal left ventricle edema on T2-weighted images, increased global relative enhancement on the T1-weighed gradient inversion recovery sequence, and epicardial late gadolinium enhancement [35]. Endomyocardial biopsies or myocardial examination at autopsy usually demonstrate multiple foci of active inflammation, focal edema, and varying degrees of myocyte degeneration or necrosis with influenza A virus occasionally isolated [37, 38].
Epidemics of influenza also are associated with an increased risk of fatal autopsy-proven myocardial infarctions, which in one study rose 30% when compared with noninfluenza winters [39].
Four children 3 to 9 years of age have been reported with myocarditis associated with the 2009 influenza A [40]. Three had fulminant myocarditis with full recovery but one died. This fatal case and one other had severe myocardial damage characterized by mononuclear infiltration [41•]. Reverse transcriptase-polymerase-chain reaction (RT-PCR) assay for influenza viral RNA was positive in cardiac tissue.
GBS Associated with Influenza or Influenza Vaccine
GBS is an uncommon disorder believed to be due to the person’s own immune system damaging peripheral nerves causing muscle weakness. Each year an estimated 6000 to 9000 cases of GBS occur in the United States. The question not fully resolved is whether influenza viral infection or influenza vaccination increases the risk for developing GBS. Epidemiologic data suggest that there is a minor temporal association with influenza-virus illnesses and developing GBS within 60 days [42]. However, the influenza-like illnesses were only clinical syndromes and not proven to be due to influenza virus making the association more tenuous. A few case reports of atypical GBS associated with influenza infection have been published during the first phase of the 2009 influenza pandemic.
In 1976, the influenza A “swine flu” vaccination program was associated with an increase of about one case of GBS per 100,000 adult vaccinations [43•]. Afterwards, a US Vaccine Adverse Event Reporting System (VAERS) supported by the US Centers for Disease Control and Prevention (CDC) was established. VAERS is a voluntary reporting system for adverse events to all vaccines including the current 2009 influenza A (H1N1) vaccine [44•]. VAERS and a similar UK reporting system have found no increased risk or a minimal increased risk of about one GBS case per million persons vaccinated associated with previous influenza vaccines [43•]. As of April 30, 2010, VAERS reported nearly 127 million doses of the 2009 vaccine being shipped to vaccination providers and having received only 136 GBS reports, which can be compared with a predicted 80 to 160 cases of GBS per week regardless of vaccination [44•]. The CDC also conducts active GBS surveillance in 10 states in their Emerging Infections Program. As of March 31, 2010, individuals who received the 2009 influenza A vaccine had an estimated incidence of 1.92 cases of GBS per 1,000,000 person-years compared with an estimated incidence for nonvaccinated individuals of 1.21 GBS cases per 100,000 person-years [45]. This significant excess risk relates to 0.8 cases per 1 million vaccinations.
Theories for the Pathogenesis of Neuromuscular Complications of Influenza Virus
It is unknown how influenza virus causes dysfunction to the human nervous system and muscles. There is abundant evidence that human influenza viruses replicate actively only in the respiratory tract. Thus, the pathogenesis for the encephalitis or myositis is unlikely to be due to widespread replication of the virus in these organs. It should be noted that in birds high-pathogenicity avian influenza A viruses do cause a viremia and replicate outside the respiratory tract in multiple organs (including the brain and muscle) often killing the bird.
A leading theory is that the respiratory viral infection triggers a cytokine cascade of proinflammatory cytokines, particularly interleukin (IL)-6 and tumor necrosis factor (TNF-α), that reach brain and muscle via blood. Studies of influenza in children with encephalopathy have reported in serum or CSF elevated levels of IL-1α, IL-6, IL-8, IL-10, IL-15, TNF-α, and soluble tumor necrosis factor receptor (TNFR1) compared with normal controls or children with mild uncomplicated influenza [41•, 46]. The highest levels of proinflammatory cytokines are seen in patients with severe encephalopathy or acute necrotizing encephalopathy and carry a poor prognosis. It is unclear how cytokines circulating in blood can cause focal lesions in brain and muscle rather than a diffuse abnormality. Of note, several studies also reported bacterial pneumonia in 30% of the severe encephalopathy patients that could contribute to the elevated cytokine levels [41•].
A second theory argues that the central nervous system dysfunction stems from a postinfectious immune-mediated process similar to a postinfectious encephalopathy seen in measles and other viruses [47]. However, the influenza encephalopathy begins early in the influenza infection and not later during the recovery phase. Molecular mimicry of influenza virus epitopes and normal brain or muscle antigens has yet to be demonstrated; however, molecular mimicry with production of antiganglioside antibodies has been postulated for the GBS peripheral neuropathy that developed after administration of the swine influenza (A/NJ/1976/H1N1) vaccine [48].
A third hypothesis suggests that the encephalopathy develops from a severe systemic respiratory infection. Because most of the children with encephalopathy develop the encephalopathy early in the infection, secondary bacterial or fungal pneumonia is unlikely the cause. It has been postulated that influenza virus releases toxic proteins that travel through the blood to the brain and muscles but such a toxic viral protein has not yet been identified for influenza virus. In addition, quantitative levels of influenza virus in the throat have not correlated with encephalopathy; however, prolonged clearance of the virus has correlated with severity of the encephalopathy [41•].
The fourth hypothesis suggests that nonpermissive or poorly replicating influenza viral infections of skeletal and myocardial fibers, endothelial cells, and hepatocytes can cause organ dysfunction. Many nonrespiratory cells are nonpermissive or defective when infected with influenza virus. In these cells the virus starts the replication cycle in the cell and causes dysfunction of the cell but fails to produce progeny virions that can produce additional cycles of replication. The nonpermissive theory argues that the initial respiratory influenza viral infection produces an early viremia that secondarily infects focal areas of brain endothelial cells (producing cerebral edema with headache or encephalopathy), myocytes (producing myalgia or myositis), myocardium (producing myocarditis), and hepatocytes (producing liver enzyme dysfunction and Reye’s syndrome).
Experimental support for the nonpermissive theory exists in experimental animals. Following intranasal inoculation, intravenous inoculation, or direct muscle injection, human influenza virus in mice or hamsters produces a nonpermissive viral infection in cerebral endothelial cells causing cerebral edema, muscle-causing myositis, myocardium-causing myocarditis, and hepatocytes causing a Reye’s syndrome-like liver dysfunction [49–51]. The nonpermissive viral infection transiently produces viral nucleic acid and viral antigens for 1 to 3 days but produces very little, if any, infectious virions and results in little tissue inflammation.
In humans, there is sporadic evidence that infection with influenza A and B viruses can cause a viremia and infect muscle, myocardium, brain, gastrointestinal tract, and liver. The evidence comes from viral isolation from damaged tissues, blood or CSF, detection of viral antigens by immunohistochemistry, and by RT-PCR assays [28, 29, 38, 52, 53]. It is difficult to know how often evidence of influenza viral infection of distant organs occurs in influenza because large clinical series have not been done. However, Fujimoto et al. [52] studied children with influenza encephalopathy and identified influenza virus by RT-PCR in CSF in 5 of 7 children. Ito et al. [53] also detected influenza virus in 1 of 9 children with influenza-associated encephalopathy.
Conclusions
The first wave of the 2009 influenza A (H1N1) infections circled the globe infecting primarily children and young adults. Accompanying the pandemic were occasional individuals who developed associated neurologic or muscular syndromes as described above. Most occurred in previously healthy children. Patients had fever, myalgias, and signs of an upper respiratory illness along with syndromes that develop early in the respiratory illness. There does not appear to be anything unusual about the respiratory infection and only occasionally did the respiratory illness progress to clinical pneumonia.
The most worrisome complication is an encephalopathy or encephalitis that occasionally rapidly progresses to acute necrotic encephalitis with lesions in both thalami and other cortical gray and white matter. The syndrome can be fatal or cause permanent sequelae. In the United States and Europe, the more typical encephalopathy may be accompanied with a generalized or focal seizure and carries a good prognosis. These children usually have normal CSF, diffuse slowing on the EEG, and normal neuroimaging. The diagnosis of the influenza-associated syndromes is made clinically with laboratory and neuroimaging supporting evidence after ruling out other etiologies. Treatment has been supportive because there is currently no evidence that administration of oseltamivir shortens the clinical course or improves outcome.
In the first wave of the pandemic, the spectrum and severity of associated neurologic and muscular syndromes were similar to those seen from influenza in prior years except that Reye’s syndrome has not been recognized. The pathogenesis of the associated syndromes remains unclear so prevention or excellent treatment approaches remain unknown.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
• Writing Committee of the WHO Consultation on Clinical Aspects Of Pandemic (H1N1) 2009 Influenza: Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med 2010, 362:1708–1719. This is an excellent current review of the clinical aspects of the 2009 influenza A pandemic.
• Centers for Disease Control and Prevention: CDC Estimates of 2009 H1N1 Influenza Cases, Hospitalizations and Deaths in the United States, April 2009–March 13, 2010. Available at http://www.flu.gov/individualfamily/about/h1n1/estimates_2009_h1n1.html. Assessed May 8, 2010. This is the most up-to-date website for US epidemiologic data on influenza available.
Miller E, Hoschler K, Hardelid P, et al.: Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010, 375:1100–1108.
Beigel JH: Influenza. Crit Care Med 2008, 36:2660–2666.
Itoh Y, Shinya K, Kiso M, et al.: In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 2009, 460:1021–1025.
Jain S, Kamimoto L, Bramley AM, et al.: Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 2009, 361:1935–1944.
Chiu SS, Tse CYC, Lau YL, Peiris M: Influenza A infection is an important cause of febrile seizures. Pediatrics 2001, 108:1004–1005.
Chung B, Wong V: Relationship between five common viruses and febrile seizures. Arch Dis Child 2007, 92:589–593.
• Toovey S: Influenza-associated central nervous system dysfunction: a literature review. Travel Med Infect Dis 2008, 6:114–124. This is an extensive literature review of neurologic complications of influenza.
Noriega LM, Verdugo RJ, Araos R, et al.: Pandemic influenza A (H1N1) 2009 with neurological manifestations, a case series. Influenza Other Respi Viruses 2010, 4:117–120.
• Centers for Disease Control and Prevention: Neurologic complications associated with novel influenza A (H1N1) virus infection in children—Dallas, Texas, May 2009. MMWR Morb Mortal Wkly Rep 2009, 58:773–778. This article presents details of four children who had seizures and encephalopathy associated with the 2009 influenza A infection.
Choi SY, Jang SH, Ihm CH, et al.: Novel swine-origin influenza A (H1N1) viral encephalitis. Yonsei Med J 2010, 51:291–292.
Lyon JB, Remigio C, Milligan T, Decline C: Acute necrotizing encephalopathy in a child with H1N1 influenza infection. Pediatr Radiol 2010, 40:200–205.
• Baltagi SA, Shoykhet M, Felmet K, et al.: Neurological sequelae of 2009 influenza A (H1N1) in children: a case series observed during a pandemic. Pediatr Crit Care Med 2010, 11:179–184. Four children had encephalopathy associated with the 2009 influenza A infection and were carefully evaluated.
Sevjar JJ, Uyeki TM: Neurologic complications of 2009 influenza A (H1N1). Neurology 2010, 74:1020–1021.
Ormitti F, Ventura E, Summa A, et al.: Acute necrotizing encephalopathy in a child during the 2009 influenza A (H1N1) pandemic: MR imaging in diagnosis and follow-up. AJNR Am J Neuroradiol 2010, 31:396–400.
German-Diaz M, Pavo-Garcia R, Diaz-Diaz J, et al.: Adolescent with neuropsychiatric symptoms associated with novel influenza A (H1N1) virus infection. Pediatr Infect Dis J 2010, 29:1–2.
Takanashi J, Tada H, Kuroki H, Barkovich AJ: Delirious behavior in influenza is associated with a reversible splenial lesion. Brain Dev 2009, 31:423–426.
Yorifuji T, Suzuki E, Tsuda T: Oseltamivir and abnormal behaviors: true or not? Epidemiology 2009, 20:619–621.
Smith JR, Sacks S: Incidence of neuropsychiatric adverse events in influenza patients treated with oseltamivir or no antiviral treatment. Int J Clin Pract 2009, 63:596–605.
Morishima T, Togashi T, Yokota S, et al.: Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin Infect Dis 2002, 35:512–517.
Lundberg A: Myalgia cruris epidemica. Acta Paediatr 1957, 46:18–31.
Rubin E, De La Rubia L, Pascual A, et al.: Benign acute myositis associated with H1N1 influenza virus infection. Eur J Pediatr 2010, 169:1159–1161.
Koliou M, Hadjiiloizou S, Ourani S, et al.: A case of benign acute childhood myositis associated with influenza A (H1N1) virus infection. Clin Microbiol Infect 2010, 16:193–195.
Congy F, Hauw JJ, Wang A, Moulias R: Influenzal acute myositis in the elderly. Neurology 1980, 30:877–878.
Panghaal V, Oritz-Romero S, Lovinsky S, Levin TL: Benign acute childhood myositis: an unusual cause of calf pain. Pediatr Radiol 2008, 38:703–705.
Kawarai T, Nishimura H, Taniguchi K, et al.: Magnetic resonance imaging of biceps femoris muscles in benign acute childhood myositis. Arch Neurol 2007, 64:1200–1201.
Gamboa ET, Eastwood AB, Hays AP, et al.: Isolation of influenza virus from muscle in myoglobinuric polymyositis. Neurology 1979, 29:1323–1335.
Kessler HA, Trenholme GM, Harris AA, Levin S: Acute myopathy associated with influenza A/Texas/1/77 infection. JAMA 1980, 243:461–462.
Dietzman DE, Schaller JG, Ray G, Reed ME: Acute myositis associated with influenza B infection. Pediatrics 1976, 57:255–258.
D’Silva D, Hewagama S, Doherty R, et al.: Novel H1N1 influenza A associated rhabdomyolysis. Pedriatr Infect Dis J 2009, 28:1138–1139.
Ayala E, Kagawa FT, Tam J, Upadhyay D: Rhabdomyolysis associated with influenza A (H1N1). JAMA 2009, 302:1863–1864.
Mamas MA, Fraser D, Neyses L: Cardiovascular manifestations associated with influenza virus infection. Int J Cardiol 2008, 130:304–309.
Karjalainen J, Nieminen MS, Heikkilä J: Influenza A1 myocarditis in conscripts. Acta Med Scand 1980, 207:27–30.
Weiss TW, Stensaeth KH, Eritsland J: Myocarditis in a juvenile patient with influenza A virus infection. Eur Heart 2010, 31:277.
Erden I, Erden EC, Ozhan H, et al.: Echocardiographic manifestations of pandemic 2009 (H1N1) influenza A virus infection. J Infect 2010, 61:60–65.
Frank H, Wittekind C, Liebert UG, et al.: Lethal influenza B myocarditis in a child and review of the literature for pediatric age groups. Infection 2010, 38:231–235.
Engblom E, Ekfors TO, Meurman OH, et al.: Fatal influenza A myocarditis with isolation of virus from the myocardium. Acta Med Scand 1983, 213:75–78.
Madjid M, Miller CC, Zarubaev VV, et al.: Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease deaths: results from 8 years of autopsies in 34,892 subjects. Eur Heart J 2007, 28:1205–1210.
El-Said HG, Bradley JS, Shayan K, et al.: Fulminant myocarditis associated with pandemic H1N1 influenza A virus in children. J Am Coll Cardiol 2010, 55:928–929.
• To KK, Hung IF, Li IW, et al.: Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis 2010, 50:850–859. This is a detailed cytokine analysis of 23 Japanese patients correlating findings with risks, severity, and prognosis.
Sivadon-Tardy V, Orlikowski D, Porcher R, et al.: Guillain-Barre syndrome and influenza virus infection. Clin Infect Dis 2009, 48:48–56.
• Haber P, Sejvar J, Mikaeloff Y, DeStanfano F: Vaccines and Guillain-Barre Syndrome. Drug Safety 2009,32:309–323. This is thorough review by the US CDC that evaluates all vaccines for an associated GBS.
• Centers for Disease Control and Prevention: Summary of 2009 Monovalent H1N1 Influenza Vaccine Data—Vaccine Adverse Event Reporting System. Available at http://vaers.hhs.gov/resources/2010H1N1Summary_May07.pdf. Assessed May 2010. This is the best website for tracking all complications of all vaccines.
Centers for Disease Control and Prevention (CDC): Preliminary results: surveillance for Guillain-Barre syndrome after receipt of influenza A (H1N1) 2009 monovalent vaccine—United States, 2009–2010. MMWR Morb Mortal Wkly Rep 2010, 59:657–661.
Ichiyama T, Morishima T, Isuma H, et al.: Analysis of cytokine levels and NF-kappaB activation in peripheral blood mononuclear cells in influenza virus-associated encephalopathy. Cytokine 2004, 27:31–37.
Johnson RT, Griffin DE, Hirsch RL, et al.: Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med 1984, 310:137–141.
Nachamkin I, Shadomy SV, Moran AP, et al.: Anti-ganglioside antibody induction by Swine (A/NJ/1976/H1N1) and other influenza vaccines: insights into vaccine-associated Guillain-Barre syndrome. J Infect Dis 2008, 198:226–233.
Davis LE, Kornfeld M, Daniels RS, Skehel JJ: Experimental influenza causes a non-permissive viral infection of brain, liver and muscle. J Neurovirol 2000, 6:529–536.
Davis LE, Kornfeld M: Experimental influenza B viral myositis. J Neurol Sci 2001, 187:61–67.
Kotaka M, Kitaura Y, Deguchi H, Kawamura K: Experimental influenza A virus myocarditis in mice. Light and electron microscopic, virologic and hemodynamic study Am J Pathol 1990, 136:409–419.
Fujimoto S, Kobayashi M, Uemura O, et al.: PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet 1998, 352:873–875.
Ito Y, Ichiyama T, Kimura H, et al.: Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza-virus-associated encephalopathy. J Med Virol 1999, 58:420–425.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davis, L.E. Neurologic and Muscular Complications of the 2009 Influenza A (H1N1) Pandemic. Curr Neurol Neurosci Rep 10, 476–483 (2010). https://doi.org/10.1007/s11910-010-0135-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-010-0135-1