
Abstract
Betula humilis is an endangered plant species in Central Europe. In order to protect this species, it is necessary to document its genetic diversity in this region and to identify areas for conservation prioritisation. As molecular investigations conducted throughout the ranges of many plant species have shown that the highest genetic diversities are found within former glacial refugia and/or within the contact zones of different phylogenetic lineages, we investigated the reasons underlying the considerable genetic variation of B. humilis in northeastern Poland revealed previously using nuclear microsatellites. We analysed 365 individuals of B. humilis from 19 populations and 67 specimens of Betula pendula and Betula pubescens sampled in four geographical regions in Poland and Belarus for polymorphism in chloroplast PCR-restriction fragment length polymorphism markers. Genetic data strongly suggested that B. humilis could have survived the Last Glacial Maximum at higher latitudes, but the hypothesis of a refugium in southeastern Poland was rejected. Chloroplast DNA analysis confirmed high genetic diversity in some populations in northeastern Poland. This phenomenon can likely be explained in terms of a suture zone, as the high haplotypic richness was followed by h T ≤ v T. Similar patterns of haplotype distributions in the birches under study and high introgression ratio (IG = 0.71) among B. humilis and congeneric trees suggested that postglacial recolonisation of the shrub birch was complicated by haplotype sharing with other birches.
Similar content being viewed by others


Introduction
Trees and shrubs of the genus Betula inhabit various ecosystems in temperate and boreal climate zones of the northern hemisphere. In the history of the Holarctic region, birches appeared in the late Cretaceous (Środoń 1979). They are rare in early tertiary sediments, but in the late tertiary, birches were common and taxonomically diverse (Němejc 1975, cited in Środoń 1979). Presently, four Betula species occur in Europe: Betula pendula Roth., Betula pubescens Ehrh., Betula nana L. and Betula humilis Schrk. Both palaeontological and genetic investigations have shown that B. pendula and B. pubescens could have survived the Last Glacial Maximum (LGM) not only in Southern European refugia but also at higher latitudes and that Holocene recolonisation occurred rapidly (Palmé et al. 2003; Willis and van Andel 2004; Maliouchenko et al. 2007; Thórsson et al. 2010). In addition, the microthermal shrub B. nana could have existed in a periglacial belt during the last glaciation (Binney et al. 2009).
Analysis of fossil fruits has shown that B. humilis was well established in northern Germany during the Weichselian (Freund et al. 2001). Macrofossils of B. humilis dated back to the Middle Pleniglacial and Late Weichselian have also been discovered in southwestern Ukraine (Starachowicz-Rybka et al. 2009). These findings strongly suggest that, like other Betula species (Palmé et al. 2003; Willis and van Andel 2004; Binney et al. 2009; Markova et al. 2009), the shrub birch could have survived the LGM at higher latitudes. However, nuclear microsatellite investigation has failed to support the hypothesis of a B. humilis refugium situated in southeastern Poland (Jadwiszczak et al. 2011a). In turn, microsatellite analysis of both sub-central B. humilis populations and populations located at the southwestern margin of the species range has revealed high genetic diversity in northeastern Poland within the area that was in close proximity to the ice sheet (Jadwiszczak et al. 2011a). Jadwiszczak et al. (2011a) suggested that this phenomenon might be explained either by survival in a periglacial area or by the mixing of different phylogenetic lineages. In order to test these hypotheses, we analysed chloroplast DNA (cpDNA) using the PCR-restriction fragment length polymorphism (RFLP) method. Organelle markers are uniparentally inherited and exhibit reduced effective population sizes; hence, they reveal a clearer picture of the genealogical history of a species than do nuclear markers (Vettori et al. 2004). Previous studies have successfully used cpDNA markers to deduce the locations of glacial refugia and postglacial colonisation routes in Alnus glutinosa (King and Ferris 1998), Fagus sylvatica (Demesure et al. 1996; Magri et al. 2006), Fraxinus excelsior (Heuertz et al. 2004), Quercus (Csaikl et al. 2002; Petit et al. 2002a, b), Populus (Cottrell et al. 2005; Fussi et al. 2010) and Betula (Palmé et al. 2003; Maliouchenko et al. 2007; Thórsson et al. 2010).
The shrub birch B. humilis is adapted to cold winters and occurs in Northeastern and Eastern Europe, as well as in a few locations in Central European countries. Birches produce light- and wind-dispersed seeds and therefore play substantial roles both in the colonisation of new areas and in the early stages of forest development. B. humilis scrub often forms natural pioneer assemblages on mesotrophic fens undisturbed by human activity. The shrub birch is endangered in Central Europe (Załuski et al. 2001). The abundance of this plant is decreasing at an alarming rate in this region due to the lowering of groundwater levels and a decline in the use of wet meadows. In order to protect B. humilis, it is necessary to document its genetic diversity and to identify areas that should be given high conservation priority.
The aims of the present study were the following: (1) to quantify the cpDNA variation of B. humilis and its geographical distribution in Poland and Belarus and (2) to examine whether this variation confirms that northeastern Poland represents an area of high genetic diversity in this species, as was previously revealed using nuclear microsatellite analysis (Jadwiszczak et al. 2011a). Because the genetic variation of B. humilis can be influenced by hybridisation with congeneric species, we also examined the pattern of cpDNA variation in the tree birches present in the same geographical regions and estimated the level of introgression between B. humilis and congeneric species in these regions.
Material and methods
Collection of samples
A total of 365 individuals of B. humilis from 19 populations were collected in four regions: northern Poland (N Poland), northeastern Poland (NE Poland), southeastern Poland (SE Poland) and Belarus. Each population was represented by 8 to 25 individuals. Names of populations, geographical positions and number of individuals per population are given in Table 1. Only specimens separated by a minimum distance of 20 m were collected in order to avoid the sampling of vegetative ramets. The collection of material in Poland was approved by the Polish Ministry of the Environment (DOPog-421-II-18.1/05/jr, DOPogiz-4211/I.A-10.3/10674/05/06, DLOPiK-op/ogiz-4211/I-83/9745/07/08/msz and DONOOŚogiz-4211/I-11/259/08/ep). B. humilis is not under protection in Belarus. Within the same four regions in which the shrub birch was sampled, we also gathered material from a total of 67 specimens of B. pendula and B. pubescens. Leaf samples were kept cold until they were transferred to the laboratory and then stored at −80°C.
Laboratory analyses
After the plant material was homogenised in liquid nitrogen, DNA was extracted using the AX Plant kit (A&A Biotechnology) according to the manufacturer’s instructions. The TF, CD and AS primer pairs designed by Taberlet et al. (1991) and Demesure et al. (1995), which amplify non-coding regions of the chloroplast genome, were chosen, because they are known to reveal genetic variation in other Betula species (Palmé et al. 2003, 2004; Maliouchenko et al. 2007). For each individual, the PCR reaction was set up in 10 μl mixture containing 3.4 μl of Multiplex PCR Master Mix (Qiagen), 2.0 μl of RNase-free water (Qiagen), 0.6 μl of primer mixture (0.2 μM of each primer) and 4.0 μl of diluted DNA. All cpDNA fragments were amplified in touchdown PCR reactions using a TProfessional thermocycler (Biometra). The TF primer program included 15 min of denaturation at 95°C, 20 cycles of 45 s at 94°C, 50 s at 51–41.5°C (−0.5°C each cycle) and 3 min of elongation at 68°C, followed by 20 cycles of 45 s at 94°C, 50 s at 41°C and 3 min at 68°C and finally 20 min at 68°C. The touchdown programs for the CD and AS primers differed from one another only in the extension temperature and included 15 min of denaturation at 95°C, 14 cycles of 45 s at 94°C, 50 s at 57–50.5°C (−0.5°C each cycle) and 4 min elongation at 68°C (AS) or 70°C (CD), followed by 18 cycles of 45 s at 94°C, 50 s at 50°C and 3 min at 68°C (AS) or 70°C (CD) and finally 20 min at 68°C (AS) or 70°C (CD). The next step was restriction. The fragments amplified by TF and CD were digested using the fast digest restriction enzymes TaqI (Fermentas) at 65°C for 10 min and HinfI (Fermentas) at 37°C for 12 min. The AS-amplified fragment was digested using only TaqI. For each digestion, 4.5 μl of PCR product was added to 9.0 μl of digestion mixture containing 7.22 μl of H2O, 0.89 μl of buffer (Fermentas) and 0.89 μl of restriction enzyme. PCR-RFLP fragments were separated by electrophoresis on horizontal 1.5 % agarose gels in 1 × TBE buffer and stained with ethidium bromide. In order to establish the size of the individual fragments, a 50-bp DNA ladder size standard (O’Range Ruler, Fermentas) was run on each gel. Gels were photographed using a UV camera (BioRad).
Data analyses
Description of haplotypes
Variation in the observed PCR-RFLP patterns is the result of either the presence or absence of a restriction site or difference in the length of the amplified fragments. We scored the polymorphism revealed by five combinations of primers and restriction enzymes (Appendix 1). In addition to variation in the length of certain fragments, we also noted some restriction sites not shared by all individuals. In one B. pubescens individual, the TF-TaqI combination revealed a new restriction site that yielded a fragment of 300 bp. Some individuals from the Sluck (SLU) population had three additional TF-HinfI restriction sites, which were visualised by fragments of 540, 500 and 280 bp. The AS-TaqI combination generated 460-bp fragments in some cases. One individual of the shrub birch from the BIA population exhibited two variants of the same AS-TaqI fragment (380 and 350 bp), indicating the appearance of a new restriction site. CD-TaqI fragments of 250 and 500 bp each occurred in single specimens from N Poland. The 240 bp CD-HinfI fragment was not observed in one individual from the Jezioro Mętne (JM) site, and a new CD-HinfI fragment of 225 bp was found in one specimen from the Dolina Rospudy (ROS) population.
Due to the presence of the unshared restriction sites, it was impossible to describe haplotypes in the manner adopted by Palmé et al. (2003). In the work conducted by Palmé et al. (2003), all haplotypes had the same restriction sites, and the only difference was in the length of the fragments. Moreover, in previous PCR-RFLP studies of other birch species (Palmé et al. 2003, 2004), the sizes of particular fragments were not compared with any size standard, which does not allow us to decode the full haplotype descriptions resulting from those studies. Hence, we developed a new system for determining haplotypes in the birches in order to enable future comparative studies (Appendix 1).
Statistical analyses
Calculation of gene diversity (H E) within each population of B. humilis and construction of the minimum spanning tree were performed using Arlequin version 3.5 (Schneider and Lischer 2009). For construction of the minimum spanning tree, the number of polymorphic sites was considered rather than the number of different fragments present in all haplotypes. For example, haplotypes I and II differed at three polymorphic sites, which resulted in the presence of six different fragments. For each geographical region and for the total sample of B. humilis, the average intra-population gene diversity (h S), the total diversity (h T) and the differentiation for unordered alleles (G ST) were calculated following Pons and Petit (1996), using the software Permut (http://www.pierroton.inra.fr/genetics/labo/Software). The corresponding parameters of diversity for ordered alleles (v S, v T, N ST) were also estimated. Permut was further used to test for phylogeographical structure (Pons and Petit 1996; Burban et al. 1999). The directly measured N ST values were compared with those obtained after 1,000 random permutations of haplotype identities, and we tested whether N ST and G ST differed significantly (Burban et al. 1999). According to Pons and Petit (1996), N ST > G ST indicates that similar haplotypes are found together in the same population more often than are randomly chosen haplotypes, suggesting that the populations are phylogeographically structured. To test for isolation by distance (IBD), the Mantel option in TFPGA version 3.1 (Miller 1997) was used. Genetic differentiation between pairs of populations was measured as F ST / (1 − F ST), and geographical distances were measured in kilometers.
As B. humilis always coexists with B. pendula and B. pubescens, an introgression ratio (IG) was estimated using the method of Belahbib et al. (2001). The introgression ratio reflects the number of locally shared haplotypes between two species, and its value ranges between 0 (when two species are totally different) and 1 (which would indicate no difference between species; Belahbib et al. 2001). The 95 % confidence interval (CI) endpoints for the IG value were calculated using the percentile bootstrap method. To achieve this, 10,000 bootstrap samples of 19 sub-populations were generated using a program (brzoza.exe) written in the C++ programming language. This program implements the Mersenne Twister pseudorandom number generator (algorithm MT19937) from the Boost Random Number Library. The source code and the compiled brzoza.exe executable are available from the corresponding author upon request. Genetic variation between B. humilis and the tree birches, B. pendula and B. pubescens, was evaluated hierarchically using analysis of molecular variance (AMOVA; Excoffier et al. 1992) in Arlequin version 3.5 (Schneider and Lischer 2009). Using 10,000 permutations, the significance of genetic variation was evaluated at three levels: between species, among populations within species and among individuals within populations.
Results
Variability of cpDNA in B. humilis
The average lengths of the PCR products were approximately 1,800 bp for TF; 3,300 bp for AS and 2,700 bp for CD. All five analysed primer-restriction enzyme pairs revealed genetic variation, and a total of 21 haplotypes were described in the three birch species (Appendix 1). The CD fragment was the most variable when cut with TaqI and HinfI, the AS was the least variable. The approximate sizes of all fragments are shown in Appendix 1.
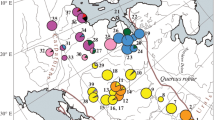
Of the 21 haplotypes described in the present study, 17 were found in B. humilis (Table 2). Haplotype I was the most abundant and widely distributed, with a frequency of 57 % in the total sample. It was found in all but two populations: ROS in NE Poland and Ozero Issa (IS) in northern Belarus (Fig. 1). Haplotype II was the next most frequent (28.5 %). It was fixed in the IS population and also appeared in nine other populations. Haplotype XIV (7.4 %) was found in five populations, including CB and Suszalewo (SUS) in NE Poland and three Belarusian populations. The fourth and final haplotype that was found in some populations was haplotype XIX (2.2 %). The remaining 13 haplotypes were private and occurred with total frequencies ranging between 0.27 and 1.37 % (Table 2). The genetic relationships among the 21 haplotypes based on the number of polymorphic sites are displayed in Fig. 2. The haplotypes formed two haplogroups in the minimum spanning tree. The first group included the common haplotype I along with 13 less frequent haplotypes, and the second group comprised haplotype II and six others. The phylogeny of the haplotypes visualised in the minimum spanning tree was congruent with their relative geographical positions.

The geographical distribution of PCR-RFLP haplotypes in B. humilis populations. Haplotype I and the related ones are marked in red and in related colors, respectively. Haplotype II and the related ones are marked in dark and light blue, respectively. Rare I rare haplotypes derived from haplotype I, Rare II rare haplotypes derived from haplotype II

Minimum spanning tree based on cpDNA haplotypes. The sizes of the circles are proportional to the haplotype frequencies. Black circles indicate the missing haplotypes. Stars indicate haplotypes found only in B. pendula and B. pubescens. The color coding is the same as in Fig. 1
The observed gene diversities (H E) varied from 0 to 0.77 (Table 1). The total diversity (h T) was average 0.57, and interpopulation differences contributed the most to this total (G ST = 0.492; Table 3). Considering each geographical region separately, the highest h T was observed in Belarus (0.804). In N Poland and NE Poland, h T values were 0.689 and 0.633, respectively. In all of these regions, h T was influenced mainly by differentiation between populations. In SE Poland, the total diversity was low (0.282), but this diversity was also generated more by interpopulation differences (G ST = 0.281) than by intra-population variation (h S = 0.203). With the exception of SE Poland, N ST was higher than G ST in all geographical regions as well as in the total sample, but this difference was statistically significant only in the total sample and in N Poland (P < 0.001 in both cases). The Mantel test revealed no isolation by distance in the study area (r = 0.0095, P = 0.952).
Variability of cpDNA in the tree birches and introgression ratio
Among the 67 individual tree birch specimens, nine haplotypes were found (Table 2). Haplotypes I and II were the most frequent, representing 53.7 and 19.4 % of the total sample, respectively. These haplotypes occurred in all geographical regions. Haplotypes VI and X were found both in NE Poland and Belarus. Among the remaining haplotypes, each appeared in one region only. Haplotypes VII, VIII, IX and XI were not observed in B. humilis.
The IG introgression ratio reached a value of 0.706 (95 % CI, 0.656–0.889). The hierarchical AMOVA revealed no differences between B. humilis and congeneric species (F CT = −0.031, P = 0.563; Table 4). Instead, most of the genetic variation was found within populations (56.68 %) and among populations within species (46.42 %).
Discussion
Geographical distribution of B. humilis haplotypes
Previous phylogeographical investigations of European birches have shown that two common haplotypes, A and C, predominate throughout the ranges of these species (Palmé et al. 2003; 2004; Maliouchenko et al. 2007), with the exception of Iceland, where another haplotype is most frequent (Thórsson et al. 2010). Although our cpDNA analysis of B. humilis was confined to Poland and Belarus, which represent the southwestern and sub-central parts of the range of this species, we obtained the same result. Within our study area, haplotypes I and II predominated in the total sample. These haplotypes are probably equivalent to the haplotypes A and C described by Palmé et al. (2003), because in both analyses, the two haplotypes differ from each other in the sizes of single AS-TaqI, CD-TaqI and CD-HinfI fragments. The most common birch haplotypes should be considered ancestral because they are centrally located in the minimum spanning tree and occur at high frequencies (Avise 2000). Polymorphisms involving more than one base substitution are likely to pre-date the LGM (Anderson et al. 2006), and hence, birch haplotypes I and II probably diverged before the last glaciation because they differ from one another by three mutations. In contrast, most of the locally occurring and rare haplotypes differ by single mutations from their ancestors, and thus, we believe that these haplotypes are the result of recent mutation events. After the last glaciation, new haplotypes also arose in A. glutinosa (King and Ferris 1998) and Quercus species (Csaikl et al. 2002).
The palynological record supports the idea that haplotypes A and C, previously described throughout the range of B. pendula, originated in western and eastern refugia, respectively (Palmé et al. 2003; Maliouchenko et al. 2007). It is very difficult to reconstruct the glacial and postglacial history of B. humilis using palaeoecological data because the macrofossil record is rather scarce, and the pollen of this species is categorised as B. nana type (Blyakharchuk et al. 2004). However, it seems possible that cold-tolerant B. humilis could have existed at higher latitudes during the LGM. This possibility is supported by multiple lines of evidence. First, fossil evidence of the shrub birch dated to the Weichselian has been found in Germany and Ukraine (Freund et al. 2001; Starachowicz-Rybka et al. 2009). The palynological record indicates that birches of the section Nanae, which includes B. humilis, could have survived the LGM in the Polish Carpathians and their foreland (Ralska-Jasiewiczowa et al. 2004). In addition, B. humilis macrofossils dated to the transition phase of the Upper Weichselian and Late Glacial periods have been detected in the Balaton Lake in Hungary (Sümegi et al. 2008). Second, present-day relict populations of the shrub birch located in Germany, Austria and Romania strongly suggest that the range of this species was much wider in the past (Załuski et al. 2001), and thus, it cannot be ruled out that B. humilis existed in both the Alps and the Carpathians during the LGM. Third, both nuclear microsatellites (Jadwiszczak et al. 2011a) and cpDNA data show no isolation by distance, which may indicate that B. humilis could have survived the LGM at relatively high latitudes. In contrast, tests of isolation by distance have revealed significant increases in among-population differentiation with the logarithm of geographic distance in F. excelsior and F. sylvatica, which also occupied Southern European refugia during the LGM (Heuertz et al. 2004; Magri et al. 2006).
It has been suggested that a refugium of B. humilis existed in SE Poland (Środoń 1979). This idea is supported neither by nuclear microsatellites (Jadwiszczak et al. 2011a) nor by our cpDNA analysis. Populations from potential glacial refugia usually exhibit high genetic diversity, and they are highly differentiated from one another due to the long period of isolation (Hewitt 1996; Hampe and Petit 2005). Microsatellite studies have shown that pairwise estimates of F ST between 18 populations of B. humilis reach their lowest values in SE Poland (Jadwiszczak et al. 2011a). A very low frequency of private alleles at nuclear microsatellite loci also contradicted the idea that SE Poland served as a refugium for the shrub birch (Jadwiszczak et al. 2011a). The present study revealed only two cpDNA haplotypes in SE Poland, and these were the most common across all study regions. This, too, undermines the hypothesis of the B. humilis glacial isolate in this region.
The most interesting result of our study was the high cpDNA variation observed in some populations of B. humilis in NE Poland. This region was in close proximity to the Scandinavian ice sheet during the LGM (Petera and Forysiak 2003), and hence, the substantial genetic diversity observed both in a previous microsatellite analysis of the shrub birch in this region (Jadwiszczak et al. 2011a) and in the present investigation was surprising. The high genetic variation in NE Poland could be explained either by the past occurrence of a glacial refugium or by the presence of a suture zone of different phylogenetic lineages. Could B. humilis have survived the LGM in NE Poland at the edge of the Scandinavian glacier? Macrofossil evidence indicates that another cold-tolerant birch species, B. nana, was located in northwestern Russia, very close to the Weichselian ice sheet (Binney et al. 2009). Taking into account the very low frequency of private alleles at nuclear microsatellite loci, Jadwiszczak et al. (2011a) argued that the presence of a suture zone of B. humilis in NE Poland was more likely. However, this issue remains unresolved. One useful tool for distinguishing a refugial area from a suture zone is the relationship between allelic richness (which is equivalent to v T) and a measure of divergence (Petit et al. 2002b; Provan and Bennett 2008). Refugium populations are characterised by high allelic richness with h T > v T, whereas high allelic richness accompanied by h T ≤ v T indicates an admixture of different genetic lineages (Petit et al. 2002b; Provan and Bennett 2008). Moreover, the allelic diversity within a refugial area is confined to one lineage, whereas the allelic diversity observed in a suture zone results in variants within multiple lineages (Petit et al. 2002b). All of the geographical regions in our study, with the exception of SE Poland, exhibited the conditions of an admixture zone. The low value of v T observed in SE Poland reflects the pattern of a newly colonised region. Hence, our analysis of cpDNA diversity supports the hypothesis that a contact zone of different phylogenetic lineages of the shrub birch exists in NE Poland. Taberlet et al. (1998) and Hewitt (1999) described five suture zones in Europe; one of these spread in Eastern Central Europe. Our result is congruent with the results of previous studies of chloroplast markers in B. pendula populations, which suggested that most of Europe was reoccupied by the waves of B. pendula migration originating near the Ural Mountains and from the north foreland of the Alps (Palmé et al. 2003; Maliouchenko et al. 2007). These two waves of migration likely came into contact and mixed in the region of Poland, forming a suture zone.
The suture zone of B. humilis revealed in the present study does not represent a sharp border between different genetic lineages. Haplotypes derived from types I and II are intermixed over several hundred kilometers. It is likely that after Holocene warming, migration waves of B. humilis spread rapidly from discrete glacial isolates and became intermixed, leading to high haplotypic diversity over a broad area in NE Poland and Belarus. Such a scenario has also been proposed for Populus tremula in Central Europe (Fussi et al. 2010) and for most populations of Salix herbacea (Alsos et al. 2009). In Quercus species, the formation of a poorly demarcated suture zone in Western Europe has been explained as the consequence of local dispersal in combination with long-distance jumps during recolonisation (Petit et al. 2002a, b). The hypothesis that B. humilis recolonised from disconnected refugia is supported by the phylogeographic structure evidenced in our total sample because N ST was significantly higher than G ST across the study area. This phylogeographic structure suggests that the study area was populated from at least two refugia (Fussi et al. 2010).
The present data do not allow us to identify the directions from which Poland and Belarus were recolonised by B. humilis. We found both of the most common haplotypes (I and II) in each geographical region. The only discrepancy between the genetic relationships and the relative geographical locations involved haplotype VI in the Belarusian SLU population. Haplotype VI is derived from haplotype II, but no specimens of haplotype II were observed in the SLU sample. One possible explanation is that haplotype VI reflects an introgression from the tree birches. The disappearance of haplotype II from the SLU location cannot be excluded, however, as this population went through a severe bottleneck in the second half of the twentieth century (see Jadwiszczak et al. 2011a). Haplotypes XIV and XIX are found solely in NE Poland and Belarus, suggesting that Eastern Europe might have been reoccupied from the east. As high frequencies of private haplotypes indicate possible locations of glacial refugia (Maggs et al. 2008), the relatively high frequency (4.2 %) of haplotype XVII found in the Belarusian SLU sample also supports the idea that a refugium of B. humilis existed in Eastern Europe. As haplotypes XIV, XVII and XIX are all related to haplotype I, this haplogroup might have come from the east. An eastern refugium has also been deduced for two other cold-tolerant plant species, Picea abies (Vendramin et al. 2000) and Pinus silvestris (Pyhäjärvi et al. 2008). In order to determine the exact locations of glacial refugia and the routes of recolonisation of B. humilis, future studies should adopt a synergistic approach involving both molecular and palaeoecological studies over the entire range of the species.
Introgression among birches
Because of haplotype sharing with other birches, the postglacial recolonisation history of B. humilis might be difficult to reconstruct. Haplotype sharing between related species typically results from one of two mechanisms: ancestral polymorphism and hybridisation/introgression (Palmé et al. 2004; Tsuda and Ide 2010). In cases where ancestral polymorphism is the sole mechanism underlying haplotype sharing between species, those species are expected to exhibit different geographical patterns of haplotype distribution (Palmé et al. 2004). For example, common haplotypes detected among Betula maximowicziana and other Japanese birches are not shared sympatrically, which suggests that these haplotypes are shared due to common ancestry (Tsuda and Ide 2010). In contrast, where shared haplotypes have resulted from frequent hybridisation/introgression, the geographical spread of these haplotypes is expected to be similar across species (Palmé et al. 2004; Tsuda and Ide 2010). Hence, a comparison of the haplotype distributions within the populations of B. humilis and those of B. pendula and B. pubescens that we sampled within four geographical regions may help us elucidate the mechanism underlying haplotype sharing in these species.
Analyses of cpDNA haplotype distributions revealed similar patterns in B. humilis and in the two congeneric tree birches that we investigated. First, haplotypes I and II were the most frequent in both B. humilis and the tree birches. Second, the third most common haplotype in B. pendula and B. pubescens was haplotype X which was found in NE Poland and Belarus. This haplotype also occurred in the SUS population of the shrub birch, which is situated in NE Poland. Third, haplotype XIV, which was detected in some stands of the shrub birch located in NE Poland and Belarus, was also found in B. pendula from Belarus. Fourth, haplotype VI was found in some of the B. pubescens samples from NE Poland and Belarus as well as in the Belarusian SLU population of B. humilis. Fifth, as in B. humilis, the highest haplotype diversities of B. pendula and B. pubescens were observed in NE Poland and Belarus. In summary, similar patterns of haplotype distributions were observed in the three birch species studied here, strongly suggesting that hybridisation/introgression processes are the main cause of haplotype sharing in these species, as previously evidenced in other European birches (Palmé et al. 2004; Thórsson et al. 2010). This idea is also supported by the value of the introgression ratio, which reflects the proportion of locally shared haplotypes. The value of IG for B. humilis and the tree birches in our study (IG = 0.71) was similar to those for B. nana/B. pubescens (IG = 0.67) and B. pubescens/B. pendula (IG = 0.79; Palmé et al. 2004).
The idea that extensive hybridisation has occurred between the shrub birch and both B. pendula and B. pubescens is also supported by the analysis of molecular variance. Our AMOVA revealed no genetic differentiation between these birch species (F CT = −0.031, P = 0.563). Similarly, haplotypic differentiation has been detected neither between B. pendula and B. pubescens nor between B. pubescens and B. nana (Palmé et al. 2004; Maliouchenko et al. 2007). Based on morphological measurements (Staszkiewicz et al. 1993) and karyotype analysis (Jadwiszczak et al. 2011c), hybridisation between B. humilis and congeneric species has previously been hypothesised to commonly occur. Comparative studies of genome sizes have also confirmed recent hybridisation events among European birches (Anamthawat-Jónsson et al. 2010).
Haplotype diversity and conservation strategies
Our analysis of three non-coding regions of cpDNA using the PCR-RFLP technique revealed 17 haplotypes in 19 populations of B. humilis. The inclusion of 36 B. pendula and 31 B. pubescens specimens in the study revealed four additional rare haplotypes. A previous study of B. pendula in 47 European localities resulted in the description of only 13 haplotypes (Palmé et al. 2003). Substantial genetic variation of B. humilis has also been revealed through the analysis of nuclear microsatellites (Jadwiszczak et al. 2011a, b). Does this mean that the endangered shrub birch exhibits higher genetic variation than does the widespread B. pendula?
In our opinion, there are three possible explanations for this unexpected pattern. The first is a difference in the sample sizes of the two species. The total sample sizes were similar (365 individuals of the shrub birch and 431 of B. pendula), but the number of samples per population was much higher for B. humilis than for B. pendula. The average sample size per population of B. humilis in our study was 19.2 individuals, whereas it was approximately half this number in the study of B. pendula (Palmé et al. 2003). Artificial selection pressure on B. pendula is a second possible explanation for the observation of fewer haplotypes in B. pendula than in B. humilis. Wood of B. pendula has long been used in the furniture and paper industries, in the manufacture of parquet and in heating homes. The harvesting of tree birches has historically led to a widespread decrease in population sizes and, consequently, to a reduction of genetic variation resulting from genetic drift. The shrub birch, which has many thin branches and reaches a height of 1–3 m, has never had any industrial value. For the present study, we chose relatively abundant populations of this species, which are expected to retain a large proportion of the former genetic variation. It has similarly been suggested that the relatively low differentiation of Quercus robur in Central Europe might also be the result of intense historical utilisation (Petit et al. 2002b). The third explanation is a more rapid replacement of generations in B. humilis, as this shrub rarely survives to the age of 20 years. Rapid replacement of generations corresponds to higher mutation rates, and it can also result in a greater probability of fixation of mutations. In Prunus spinosa, another European wild shrub, high total cpDNA diversity is also potentially explained by a lack of selection pressure and a high generational turnover (Mohanty et al. 2000).
In order to maintain the current level of genetic variation of the endangered B. humilis, active protection is required. The main obstacle to the conservation of the shrub birch is the overgrowing of its habitats by forest plants (Załuski et al. 2001). To avoid this, coppicing should be regularly conducted. During the few years of our study, coppicing was conducted at the JM locality in N Poland. We observed the appearance of many new branches of B. humilis in this previously declining population, and this population exhibited the highest haplotype diversity of all populations in that region. As the Polish populations of B. humilis constitute the southwestern margin of the continuous range of this species, their protection can prevent the extinction of the shrub birch in Central Europe. Based on nuclear microsatellite analyses, Jadwiszczak et al. (2011a) identified two groups of B. humilis populations that are particularly worthy of protection. The first group includes the TM and JM populations in N Poland, and the second group includes the populations in the northeastern part of the country. The present study confirms both the uniqueness of the JM sample, where private haplotypes III, IV, and V are found, and the high genetic diversity in NE Poland, and hence, we confirm that B. humilis populations from N Poland and from NE Poland are the most valuable units for conservation. Conservation of the shrub birch is important for the maintenance of peatland biodiversity. Peatlands usually represent relatively low biological diversity; hence, the extinction of one species could cause disastrous changes in this environment (Minayeva et al. 2009). Reciprocally, the existence of peatlands and fens is necessary to protect B. humilis and other rare and endangered species for which this type of habitat is the last refuge (Minayeva et al. 2009).
References
Alsos IG, Alm T, Normand S, Brochmann C (2009) Past and future range shifts and loss of diversity in dwarf willow (Salix herbacea L.) inferred from genetics, fossils and modelling. Global Ecol Biogeogr 18:223–239. doi:10.1111/j.1466-8238.2008.00439.x
Anamthawat-Jónsson K, Thórsson ÆTh, Temsch EM, Greilhuber J (2010) Icelandic birch polyploids—the case of a perfect fit in genome size. J Bot, article ID 347254: 9 pages. doi:10.1155/2010/347254
Anderson LL, Hu FS, Nelson DM, Petit RJ, Paige KN (2006) Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. Proc Natl Acad Sci USA 103:12447–12450. doi:10.1073/pnas.0605310103
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Belahbib N, Pemonge MH, Ouassou A, Sbay H, Kremer A, Petit RJ (2001) Frequent cytoplasmic exchange between oak species that are not closely related: Quercus suber and Q. ilex in Morocco. Mol Ecol 10:2003–2012. doi:10.1046/j.0962-1083.2001.01330.x
Binney HA, Willis KJ, Edwards ME, Bhagwat SA, Anderson PM, Andreev AA, Blaauw M, Damblon F, Haesaerts P, Kienast F, Kremenetski KV, Krivonogov SK, Lozhkin AV, MacDonald GM, Novenko EY, Oksanen P, Sapelko TV, Väliranta M, Vazhenina L (2009) The distribution of late–Quaternary woody taxa in northern Eurasia: evidence from a new macrofossil database. Quatern Sci Rev 28:2445–2464. doi:10.1016/j.quascirev.2009.04.016
Blyakharchuk TA, Wright HE, Borodavko PS, van der Knaap WO, Ammann B (2004) Late Glacial and Holocene vegetational changes on the Ulagan high-mountain plateau, Altai Mountains, southern Siberia. Palaeogeogr Palaeoclimatol Palaeoecol 209:259–279. doi:10.1016/j.palaeo.2004.02.011
Burban C, Petit RJ, Carcreff E, Jactel H (1999) Rangewide variation of the maritime pine bast scale Matsucoccus feytaudi Duc. (Homoptera: Matsucoccidae) in relation to the genetic structure of its host. Mol Ecol 8:1593–1602. doi:10.1046/j.1365-294x.1999.00739.x
Cottrell JE, Krystufek V, Tabbener HE, Milner AD, Connolly T, Sing L, Fluch S, Burg K, Lefèvre F, Achard P, Bordács S, Gebhardt K, Vornam B, Smulders MJM, Vanden Broeck AH, Slycken JV, Storme V, Boerjan W, Castiglione S, Fossati T, Alba N, Agúndez D, Maestro C, Notivol E, Bovenschen J, van Dam BC (2005) Postglacial migration of Populus nigra L.: lessons learnt from chloroplast DNA. Forest Ecol Manag 206:71–90. doi:10.1016/j.foreco.2004.10.052
Csaikl UM, Glaz I, Baliuckas V, Petit RJ, Jensen JS (2002) Chloroplast DNA variation of white oak in the Baltic countries and Poland. Forest Ecol Manag 156:211–222
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Demesure B, Comps B, Petit RJ (1996) Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution 50:2515–2520
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA. Genetics 131:479–491
Freund H, Birks HH, Birks HJB (2001) The identification of wingless Betula in Weichselian sediments in Gross Todtshorn borehole (Lower Saxony, Germany)—the occurrence of Betula humilis Schrank. Veg Hist Archaeobot 10:107–115. doi:10.1007/PL00006919
Fussi B, Lexer C, Heinze B (2010) Phylogeography of Populus alba (L.) and Populus tremula (L.) in Central Europe: secondary contact and hybridization during recolonisation from disconnected refugia. Tree Genet Genomes 6:439–450. doi:10.1007/s11295-009-0262-5
Hampe A, Petit R (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467. doi:10.1111/j.1461-0248.2005.00739.x
Heuertz M, Fineschi S, Anzidei M, Pastorelli R, Salvini D, Paule L, Frascaria-Lacoste N, Hardy OJ, Vekemans X, Vendramin GG (2004) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13:3437–3452. doi:10.1111/j.1365-294X.2004.02333.x
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. doi:10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112. doi:10.1111/j.1095-8312.1999.tb01160.x
Jadwiszczak KA, Banaszek A, Jabłońska E, Sozinov OV (2011a) Could Betula humilis Schrk. have survived the last glaciation at a current margin of its distribution?—testing the hypothesis of glacial refugium using nuclear microsatellites. Plant Syst Evol 297:147–156. doi:10.1007/s00606-011-0503-6
Jadwiszczak KA, Jabłońska E, Banaszek A (2011b) Genetic diversity of the shrub birch Betula humilis Schrk. at the south-western margin of its range. Plant Biosyst. doi:10.1080/11263504.2011.557100
Jadwiszczak KA, Jabłońska E, Kłosowski S, Banaszek A (2011c) Aneuploids in the shrub birch Betula humilis populations in Poland. Acta Soc Bot Pol 80:233–235. doi:10.5586/asbp. 2011.015
King RA, Ferris C (1998) Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol Ecol 7:1151–1161
Maggs CA, Castilho R, Foltz D, Henzler C, Taimour Jolly M, Kelly J, Olsen J, Perez KE, Stam W, Väinölä R, Viard F, Wares J (2008) Evaluating signatures of glacial refugia for north Atlantic benthic marine taxa. Ecology 89(Supplement):108–122
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latałowa M, Litt T, Paule L, Roure JM, Tantau I, van der Knaap WO, Petit RJ, de Beaulieu JL (2006) A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171:199–221. doi:10.1111/j.1469-8137.2006.01740.x
Maliouchenko O, Palmé AE, Buonamici A, Vendramin GG, Lascoux M (2007) Comparative phylogeography and population structure of European Betula species, with particular focus on B. pendula and B. pubescens. J Biogeogr 34:1601–1610. doi:10.1111/j.1365-2699.2007.01729.x
Markova AK, Simakova AN, Puzachenko AY (2009) Ecosystems of Eastern Europe at the time of maximum cooling of the Valdai glaciation (24–18 kyr BP) inferred from data on plant communities and mammal assemblages. Quatern Int 201:53–59. doi:10.1016/j.quaint.2008.05.020
Miller MP (1997) Tools for population genetic analyses (TFPGA) version 1.3: a Windows program for the analysis of allozyme and molecular population genetic data. Computer software distributed by author
Minayeva T, Sirin A, Bragg O (2009) A quick scan of peatlands in Central and Eastern Europe. Wetlands International, Wageningen, pp 1–132
Mohanty A, Martín JP, Aguinagalde I (2000) Chloroplast DNA diversity within and among populations of the allotetraploid Prunus spinosa L. Theor Appl Genet 100:1304–1310. doi:10.1007/s001220051439
Palmé AE, Su Q, Rautenberg A, Manni F, Lascoux M (2003) Postglacial recolonization and cpDNA variation of silver birch, Betula pendula. Mol Ecol 12:201–212
Palmé AE, Su Q, Palsson S, Lascoux M (2004) Extensive sharing of chloroplast haplotypes among European birches indicates hybridization among Betula pendula, B. pubescens and B. nana. Mol Ecol 13:167–178. doi:10.1046/j.1365-294X.2003.02034.x
Petera J, Forysiak J (2003) The problem of the last glaciation extent in central Poland. Geol Quart 47:357–366
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam B, Deans JD, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Jensen JS, König AO, Lowe AJ, Madsen SF, Mátyás G, Munro RC, Popescu F, Slade D, Tabbener H, de Vries SGM, Ziegenhagen B, de Beaulieu JL, Kremer A (2002a) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. Forest Ecol Manag 156:49–74
Petit RJ, Csaikl UM, Bordács S, Burg K, Coart E, Cottrell J, van Dam B, Deans JD, Dumolin-Lapègue S, Fineschi S, Finkeldey R, Gillies A, Glaz I, Goicoechea PG, Jensen JS, König AO, Lowe AJ, Madsen SF, Mátyás G, Munro RC, Olalde M, Pemonge MH, Popescu F, Slade D, Tabbener H, Taurchini D, de Vries SGM, Ziegenhagen B, Kremer A (2002b) Chloroplast DNA variation in European white oaks. Phylogeography and patterns of diversity based on data from over 2600 populations. Forest Ecol Manag 156:5–26
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Provan J, Bennett KD (2008) Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol 23:564–571. doi:10.1016/j.tree.2008.06.010
Pyhäjärvi T, Salmela MJ, Savolainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4:247–254. doi:10.1007/s11295-007-0105-1
Ralska-Jasiewiczowa M, Wacnik A, Mamakowa K, Nalepka D (2004) Betula L.—Birch. In: Ralska-Jasiewiczowa M, Latałowa M, Wasylikowa K, Tobolski K, Madeyska E, Wright HE Jr, Turner C (eds) Late Glacial and Holocene history of vegetation in Poland based on isopollen maps. W. Szafer Institute of Botany, PAN, Kraków, pp 57–68
Schneider S, Lischer H (2009) Arlequin, version 3.5: an integrated software package for population genetics data analysis. University of Berne, Berne
Środoń A (1979) Birch in the past. In: Białobok S (ed) Birches. PWN, Warszawa–Poznań, pp 9–23, in Polish with English summary
Starachowicz-Rybka R, Granoszewski W, Hrynowiecka-Czmielewska A (2009) Quaternary environmental changes at Starunia palaeontological site and vicinity (Carpathian region, Ukraine) based on palaeobotanical studies. Ann Soc Geol Pol 79:279–288
Staszkiewicz J, Białobrzeska M, Truchanowicz J, Wójcicki JJ (1993) Variability of Betula humilis (Betulaceae) in Poland. 4. Hybrid and introgressive forms. Frag Flor Geobot 38:475–488
Sümegi P, Gulyás S, Jakab G (2008) Holocene paleoclimatic and paleohydrological changes in Lake Balaton as inferred from a complex quantitative environmental historical study of a lacustrine sequence of the Szigliget embayment. Documenta Praehistorica XXXV:33–43
Taberlet P, Gielly L, Patou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Taberlet P, Fumagali L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Thórsson ÆTh, Pálsson S, Lascoux M, Anamthawat-Jónsson K (2010) Introgression and phylogeography of Betula nana (diploid), B. pubescens (tetraploid) and their triploid hybrids in Iceland inferred from cpDNA haplotype variation. J Biogeogr 37:2098–2110. doi:10.1111/j.1365-2699.2010.02353.x
Tsuda Y, Ide Y (2010) Chloroplast DNA phylogeography of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in Japan. J Plant Res 123:343–353. doi:10.1007/s10265-009-0280-2
Vendramin GG, Anzidei M, Madaghiele A, Sperisen C, Bucci G (2000) Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 43:68–78. doi:10.1139/g99-093
Vettori C, Vendramin GG, Anzidei M, Pastorelli R, Paffetti D, Giannini R (2004) Geographic distribution of chloroplast variation in Italian populations of beech (Fagus sylvatica L.). Theor Appl Genet 109:1–9. doi:10.1007/s00122-004-1609-9
Willis KJ, van Andel TH (2004) Trees or no trees? The environments of central and eastern Europe during the Last Glaciation. Quatern Sci Rev 23:2369–2387. doi:10.1016/j.quascirev.2004.06.002
Załuski T, Pisarek W, Kucharczyk M, Kamińska AM (2001) Betula humilis Schrank. In: Kaźmierczakowa R, Zarzycki K (eds) Polish plant red book. W. Szafer Institute of Botany, PAN, Kraków, pp 79–81 (in Polish with English summary)
Acknowledgements
We thank S. Kłosowski, E. Żuk-Kempa, K. Wasilewska, R. Syty and P. Jadwiszczak for their help in the field and laboratory. Special thanks go to I. Święcicka and T. Hauschild for the access to electrophoretic laboratory. We are grateful to two anonymous reviewers for their insightful and constructive comments on the earlier draft of the manuscript. This research was supported by a grant from the Polish Ministry of Science and Higher Education (grant no. NN303 3763 33).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Aitken
Appendix 1
Appendix 1
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jadwiszczak, K.A., Banaszek, A., Jabłońska, E. et al. Chloroplast DNA variation of Betula humilis Schrk. in Poland and Belarus. Tree Genetics & Genomes 8, 1017–1030 (2012). https://doi.org/10.1007/s11295-012-0482-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-012-0482-y