
Abstract
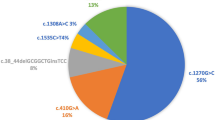
Early recognition by newborn screening and oral biotin supplementation may prevent clinical and neurological deficits in profound biotinidase deficiency (residual plasma biotinidase activity <10%). In order to evaluate possible correlations of molecular characteristics, onset and continuation of treatment and clinical outcome, we investigated 21 patients detected by newborn screening and consecutive family investigations. In 18 patients found by newborn screening, the range of biotinidase activities was 0%–9% residual activity. Application of a sensitive HPLC assay enabled us to discriminate five patients with residual biotinidase activities <1%. Two patients with zero activities were homozygous for the G98:d7i3 mutation and three patients with activities <1% carried mutations G98:d7i3, R157H, and Q456H. The mutation spectrum of the remaining patients included T532M, A171T+D444H, V62M,C432W, and D444H. Evaluation of clinical and neuropsychological outcome showed that only patients with biotinidase activities <1% exhibited characteristic clinical symptoms within the first weeks of life whereas five patients with residual activities of 1.2%–4.6% did not develop clinical symptoms even when not treated until 3.5–21 years. In all patients treated with biotin within the first weeks of life, neuropsychological outcome was normal whereas abnormal in three out of five patients tested for IQ and treated after the age of 3.5 years. Conclusion:the clinical and molecular spectrum of profound biotinidase deficiency is heterogeneous. Early onset of symptoms is predicted by the presence of zero residual activity as measured by sensitive assays and by homozygosity for the G98:d7i3 mutation. In patients with higher residual activities and variable mutational spectrum, correlation with the onset and severity of symptoms cannot be made.
Similar content being viewed by others


Abbreviations
- BD :
-
biotinidase deficiency
- DGGE :
-
denaturing gradient gel electrophoresis
References
Baumgartner ER, Suormala T (1997) Multiple carboxylase deficiency: inherited and acquired disorders of biotin metabolism. Int J Vitam Nutr Res 67: 377–384
Cole H, Reynolds TR, Lockyer JM (1994) Human serum biotinidase: cDNA cloning sequence and characterisation. J Biol Chem 269: 6566–6570
Cole-Knight H, Reynolds TR, Meyers GA, Pompiono RJ, Buck GA, Wolf B (1998) Structure of the human biotinidase gene. Mamm Genome 9 327–330
Dilling H, Mombour W, Schmidt MH, Schulte-Markwort E (1993) Weltgesundheits-organisation. Internationale Klassifikation psychischer Störungen. ICD 10 Kapital V (F). Klinisch–diagnostische Leitlinien, 2. Auflage. Huber H Verlag, Bern Göttingen
Frigg M, Brubacher G (1976) Biotin deficiency in chicks fed a wheat–based diet. Int J Vit Nutr Res 46: 314–321
Heard GS, Secor-McVoy JR, Wolf B (1984) A screening method for biotindase deficiency in newborns. Clin Chem 3: 125–127
Möslinger D, Stöckler–Ipsiroglu S, Scheibenreiter S, Tiefenthaler M, Mühl A, Seidl R, Strobl W, Plecko B, Suormala T, Baumgartner R (2001) Clinical and neuropsychological outcome in 33 patients with biotinidase deficiency ascertained by nationwide newborn screening and family studies in Austria. Eur J Pediatr 160: 277–282
Mühl A, Möslinger D, Item CB, Stöckler–Ipsiroglu S (2001) Molecular characterisation of 34 patients with biotinidase deficiency ascertained by newborn screening and family investigation. Eur J Hum Genet 9: 237–243
Norrgard KJ, Pompiono RJ Swango KL, Hymes J, Reynolds TR, Buck GA, Wolf B (1997) Mutation Q456H is the most common cause of profound biotindase deficiency in children ascertained by newborns screening in the United States. Biochem Mol Med 6: 22–27
Norrgard KJ, Pompiono RJ, Hymes J, Wolf B (1999) Mutations causing profound biotinidase deficiency in children ascertained by newborns screening in the United States occur at different frequencies than in symptomatic children. Pediatr Res 46: 20−27
Pompiono RJ, Hymes J, Reynolds TR, Meyers GA, Fleischhauer K, Buck GA, Wolf B (1997) Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis. Pediatr Res 42: 840–848
Suormala T, Baumgartner ER, Wick H, Scheibenreiter S, Schweitzer S (1990) Comparison of patients with complete and partial biotindase deficiency: biochemical studies. J Inherit Metab Dis 13: 76–92
Suormala T, Ramaekers VT, Schweitzer S, Fowler B, Laub MC, Schwermer C, Bachmann J, Baumgartner ER (1995) Biotinidase Km variants: detection and detailed biochemical investigations. J Inherit Metab Dis 18: 689–700
Wolf B (2001) Disorders of biotin metabolism. In Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited disease. McGraw–Hill, New York, pp 3935–3960
Wolf B, Norrgard D, Pompiono RJ, Mock DM, Secor-McVoy JR, Fleischhauer K, Shapiro S, Blitzer MG, Hymes J (1997) Profound biotinidase deficiency in two asymptomatic adults, Am J Med Genet 73: 5–9
Wolf B, Pompiono RJ, Norrgard KJ, Lott IT, Baumgartner R, Suormala T, Ramaekers VT, Coscun T, Tokatli A, Ozalp I, Hymes J (1998) Delayed onset of profound biotinidase deficiency. J Pediatr 132: 262–265
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Möslinger, D., Mühl, A., Suormala, T. et al. Molecular characterisation and neuropsychological outcome of 21 patients with profound biotinidase deficiency detected by newborn screening and family studies. Eur J Pediatr 162 (Suppl 1), S46–S49 (2003). https://doi.org/10.1007/s00431-003-1351-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-003-1351-3