
Abstract.
The parvins are a family of proteins involved in linking integrins and associated proteins with intracellular pathways that regulate actin cytoskeletal dynamics and cell survival. Both α-parvin (PARVA) and β-parvin (PARVB) localize to focal adhesions and function in cell adhesion, spreading, motility and survival through interactions with partners, such as integrin-linked kinase (ILK), paxillin, α-actinin and testicular kinase 1. A complex of PARVA with ILK and the LIM protein PINCH-1 is critical for cell survival in a variety of cells, including certain cancer cells, kidney podocytes and cardiac myocytes. While PARVA inhibits the activities of Rac1 and testicular kinase 1 and cell spreading, PARVB binds αPIX and α-actinin, and can promote cell spreading. In contrast to PARVA, PARVB inhibits ILK activity and reverses some of its oncogenic effects in cancer cells. This review focuses on the structure and function of the parvins and some possible roles in human diseases.
Similar content being viewed by others


Introduction
The actin cytoskeleton plays a critical role in maintaining cellular shape, regulating cell motility and responding to mechanical strain. It is highly dynamic and responds to extracellular stimuli, such as hormones and growth factors, cell-cell adhesion and variations in the extracellular matrix (ECM). Cell-ECM adhesion is mediated primarily by integrins and associated proteins. Integrins function as transmembrane receptors for ECM proteins such as fibronectin, laminin and collagen. Through protein complexes associated with their cytoplasmic domain, such as those containing talin, integrin-linked kinase (ILK), PINCH, parvin, paxillin and/or focal adhesion kinase (FAK), integrins transduce bi-directional signals between the ECM and intracellular signaling pathways. Integrin-associated proteins can integrate cell adhesion-mediated signaling with other extracellular signals, such as those originating from growth factor receptors, through a variety of intracellular signaling pathways. In addition to regulating cell survival, proliferation and gene expression, integrin-mediated signaling often results in changes in the actin cytoskeleton leading to cell shape change and motility. In this article, we will review current knowledge about a family of proteins, the parvins, which play a critical role in transducing signals from integrins to the actin cytoskeleton and intracellular signaling proteins.
Structural features
The mammalian parvin protein family has three members (α-, β- and γ-parvin). They were identified independently at approximately the same time by several laboratories based on their protein binding activities or sequence homology. Approximately 5 years ago, Nikolopoulos and Turner [1] identified and cloned a new F-actin and paxillin LD1 motif binding protein from a rat complementary DNA (cDNA) library and named it actopaxin. In a yeast two-hybrid screening of ILK binding partners, Tu et al. [2] identified and cloned a new ILK binding protein that contains two CH domains and hence named it Calponin Homology domain-containing ILK Binding Protein or CH-ILKBP. In an independent yeast two hybrid screening, Yamaji et al. [3] identified and cloned another new protein that interacts with ILK, which they named affixin. Based on sequence homology with the actin binding domain of α-actinin, Olski et al. [4] identified and cloned three structurally related proteins and named them α-, β- and γ-parvin, respectively. A comparison of the DNA and protein sequences shows that actopaxin and CH-ILKBP are identical to the alpha member and affixin is identical to the beta member of the parvin family. The members of the parvin family are encoded by three different genes (parva, parvb and parvg) in mammalian organisms. Proteins that are structurally and functionally related to parvins have been identified in invertebrates such as Caenorhabditis elegans by sequence analyses [4] and independent genetic studies [5]. Thus, Parvin appears to be an ancient protein family and is well conserved during evolution.
Human PARVA, PARVB and PARVG contain 372, 364 and 331 amino acids (with theoretical molecular masses of 42,330, 41,770 and 37,485), respectively (fig. 1). In addition, PARVB transcript has two alternative initiation sites, which result in proteins of 350 and 313 amino acids (with theoretical molecular masses of 40,130 and 35,924). The shorter forms of PARVB are herein respectively abbreviated as PARVB-s and PARVB-ss in contrast with full-length PARVB-l (fig. 1). In human rhabdomyosarcoma cells, an additional alternatively spliced isoform of PARVB was identified, named CLINT [6]. This isoform originates from an alternative upstream exon coding for 70 amino acids and results in a protein with 397 amino acids (fig. 1). For a comprehensive study of the genomic organization of the parvins see [7].

Structural features of PARVA and PARVB proteins. Numbers indicate amino acid positions. Protein binding sites are indicated with a horizontal line. Effect of point mutations is indicated by ↓. Sequences required for localization are indicated by ↑. Both PARVA and PARVB contain two calponin homology domains (CH) and nuclear localization sequences (NLS) at their N-termini. In addition to full-length PARVB (PARVB-l), PARVB-s and PARVB-ss can be produced by alternative translation initiation. A PARVB isoform expressed from alternative 5′ exons (CLINT) has also been detected in certain cell types.
Human PARVA and PARVB share 74% identity and 85% similarity, whereas the more distantly related PARVG shares 42% identity and 67% similarity with PARVA. The most distinct feature of parvins is the presence of two calponin homology (CH) domains in their C-terminal regions, named CH1 and CH2 (fig. 1). Each CH domain comprises approximately 100 amino acids, separated by a 60-amino acid linker. These CH domains are considered ‘atypical’ in that they do not exactly match the consensus sequences of type 1 and type 2 CH domains present in a large number of actin binding proteins such as α-actinin, spectrin and filamin [4, 8]. The parvin CH1 domain is more related to the type 1 CH domain of spectrin, whereas the second CH domain is more related to the type 1 CH domain of α-actinin [4, 8]. The CH1 and CH2 domains of the parvins are tentatively classified as type 4 and type 5 CH domains, respectively [8]. In addition to the conserved CH domains, the parvins have a 60-amino acid linker with some homology to regions preceding the CH1 of β-spectrin and plectin, as well as N-terminal extensions that show a high degree of variability between the various parvins, from 82 amino acids in PARVG to 125 amino acids in PARVB and 133 amino acids in PARVA (fig. 1). Both PARVA and PARVB have nuclear localization signals in the N-terminal extensions, as well as src-homology SH3 binding sites of the consensus PXXP, where X is any amino acid but cysteine [4].
Tissue distribution
In mammalian organisms, PARVA and PARVB are ubiquitously expressed but enriched in heart and skeletal muscle (table 1). In the spleen and platelets [9], the predominant isoform of PARVB is a short form translated from a downstream initiation codon, PARVB-ss (fig. 1). PARVG has a more restricted tissue distribution, as determined from messenger RNA (mRNA) blots, with predominance of lymphoid and hematopoietic tissues [7]. Since currently there are no published studies addressing the functions of PARVG, we will focus on PARVA and PARVB in our discussions.
Parvin interactions
Parvin proteins do not possess intrinsic catalytic activities and therefore their functions are mediated through interactions with various binding partners. One of the key parvin binding partners is ILK [10–13] (fig. 2). PARVA and PARVB have clearly been demonstrated to interact with ILK by different binding assays including yeast two-hybrid, co-immunoprecipitation and glutathione Stransferase (GST) fusion pull-down assays [2, 3, 14]. The ILK binding site has been mapped to the CH2 domain of PARVA and PARVB. F271 within this domain appears to be critical for the ILK binding activity of PARVA, as a single point mutation at this position (F271D) abolished its interaction with ILK [2]. Although the CH2 domain is sufficient for interacting with ILK, residues located outside of the CH2 domain can modulate the interaction with ILK. The N-terminus of PARVA contains multiple proline-directed Ser/Thr phosphorylation sites [15]. Deletion of 22 residues from the N-terminus of PARVA inhibits the proline-directed Ser/Thr phosphorylation of PARVA and reduces its complex formation with ILK [16].

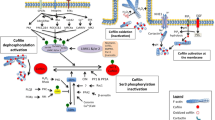
A working model for the functions of parvins and their binding proteins. ILK binds PINCH through its N-terminal domain and binds PARVA or PARVB through its C-terminal domain, resulting in formation of PINCH-ILK-parvin ternary complexes. Formation of these complexes occurs before localization to integrin-rich adhesion sites and is required for preventing degradation of ILK, PINCH1 and parvins. Recruitment of these complexes to integrin-rich adhesion sites is mediated by interactions of the complexes with additional proteins, such as β1-integrin, Mig-2 and paxillin. Phosphorylation of the N-terminus of PARVA increases binding to ILK and inhibits binding to TESK-1. Since PARVA inhibits TESK-1 activity, release of TESK-1 upon phosphorylation of PARVA results in inhibition of cofilin by phosphorylation and consequently, reduction of the turnover rates of actin filaments. PARVA can bind to F-actin in vitro but the functional significance of this interaction remains to be established. The PINCH1-ILK-PARVA complex plays a critical role in cell survival by promoting membrane recruitment of Akt and its activation by phosphorylation. PARVA and PARVB compete for binding to ILK and have opposite effects on ILK kinase activity. While PARVA binding increases ILK kinase activity, PARVB binding represses it. Also, PARVB promotes adhesion and spreading through binding of α-actinin, α-PIX and activation of Rac1, while PARVA represses Rac1.
On the other side of the binding interface, the parvin binding site has been mapped to the C-terminal kinase domain of ILK [2, 14]. The binding of PARVA and PARVB to ILK is mutually exclusive [17], suggesting that a single CH2-binding site in the ILK C-terminal domain mediates the interactions with PARVA and PARVB. ILK contains a PH-like motif that partially overlaps with the C-terminal kinase domain. The PH-like motif can interact with certain phospholipids such as PIP3, which is produced by the action of PI3-kinase. The PI3-kinase signaling pathway appears important for regulation of the ILK-PARVA interaction, since PI3-kinase inhibition or overexpression of the PIP3 phosphatase PTEN can reduce the interaction [18]. However, deletion of amino acid residues 180–190 within the PH-like motif does not inhibit the PARVA interaction [19].
In addition to binding PARVA or PARVB through the C-terminal domain, ILK interacts with PINCH-1 and PINCH-2, members of a family of focal adhesion proteins that consist of five LIM domains, through the N-terminal ankyrin repeat domain of ILK [19–21]. Through these interactions, PINCH, ILK and parvin proteins form ternary complexes in cells (fig. 2). The formation of the PINCH-ILK-parvin complexes occurs prior to their localization to cell-matrix adhesions [19]. One important consequence of the formation of the PINCH-ILKparvin complexes is the stabilization of these proteins by protecting them from proteosomal degradation [22]. Furthermore, formation of the PINCH-ILK-parvin complexes facilitates their localization to cell-matrix adhesions. Interestingly, treatment of cells with calphostin C dramatically inhibits the formation of the PINCH-ILKparvin complexes [19], suggesting a role of protein kinase C (PKC) or other calphostin C-responsive signaling pathways in this process.
Another interaction mediated by PARVA is that with paxillin, an important component of focal adhesions [23, 24]. Nikolopoulos et al. have mapped the paxillin binding site to amino acids 273–290 of PARVA and the PARVA binding sites to LD1 and LD4 motifs of paxillin [1, 14]. Interestingly, paxillin also interacts with ILK through the LD1 motif [14]. The interactions of paxillin with ILK and PARVA likely contribute to the localization of the PINCH-ILK-parvin complexes to focal adhesions.
In addition to interacting with ILK and paxillin, PARVA can interact with F-actin [1, 4]. PARVA binds to F-actin with an affinity that is comparable to those of many other actin binding proteins (Kd = 8.4 µM) [4]. Detailed characterization of the F-actin binding sites in the CH domains of PARVA remains to be done.
Recently, Lalonde et al. [25] showed that PARVA binds to testicular protein kinase 1 (TESK1). TESK1 can phosphorylate cofilin and promote F-actin polymerization and cell spreading [26–28]. Interestingly, PARVA binds to TESK1 and inhibits its activity [25]. RNA interference (RNAi)-mediated depletion of PARVA promotes cell spreading [17]. PARVA likely suppresses cell spreading by regulating multiple signaling pathways, including suppression of Rac1 activation [17] and inhibition of TESK1 [25].
As mentioned above, PARVB, like PARVA, interacts with ILK. However, despite the structural similarity and extensive efforts, PARVB has not been demonstrated to bind to actin or paxillin [3, 29]. Yamaji et al. [29] recently demonstrated that PARVB interacts with α-actinin via the CH2 domain. In addition, PARVB interacts with αPIX [30], a guanine nucleotide exchange factor (GEF) for Rac1 and possibly other Rho GTPases, through its CH1 domain. PARVB likely functions in actin cytoskeletal remodeling and cell spreading via interactions with multiple partners, including ILK, α-actinin and αPIX.
Subcellular Localization
When cells attach to the ECM, several changes occur at the membrane and cytoskeleton [31, 32]. First, nascent cell-ECM contacts (focal complexes) are established, where integrins engage with the ECM and recruit proteins, such as talin, paxillin, PINCH, ILK, parvins, α-actinin, vinculin, FAK and VASP. This leads to recruitment of additional F-actin and F-actin associated proteins and maturation into focal adhesions (FAs). In addition to the establishment of FAs, spreading cells will extend wide membrane protrusions, called lamellipodia, also present in some cells initiating directional migration. Soon after identification of parvins as ILK and paxillin binding proteins, it became apparent that protein-protein interactions of parvins with these partners were critical for their subcellular localization. More specifically, the formation of complexes of ILK with parvins and the LIM proteins PINCH-1 and PINCH-2 became a paradigm for multiprotein complex formation, stabilization and localization in the context of cell adhesion.
Both PARVA and PARVB, together with ILK, are observed in lamellipodia in spreading cells, although it appears that ILK, PARVB and PINCH-1 are expressed very early at the plasma membrane during lamellipodia extension before FAK, vinculin and paxillin are recruited [3, 20]. Established FAs clearly contain both PARVA and PARVB. In addition to FAs and lamellipodia, ILK, PINCH and PARVA staining is observed in a fibrillar pattern more intense towards the center of the cells [1, 4, 19, 33]. PARVA-containing central fibers correspond to tensin-containing fibrillar adhesions [4]. Tensin-containing fibrillar adhesions are highly dynamic structures that overlay fibronectin fibrils in the ECM and move towards the center through the action of actomyosin [31, 32]. Localization of PARVA to epithelial cell-cell adhesion junctions, rich in cadherins and catenins, has been observed in some epithelial cells [4] but not in others [2, 34], possibly indicating some cell-type specificity. PARVB has been found associated with thin stress fibers (involved in lamellipodia protrusion), where it co-localizes with α-actinin, but is excluded from mature stress fibers (involved in maintaining cell architecture), where α-actinin crosslinks actin filaments [29].
The domains responsible for the various subcellular localizations of PARVA and PARVB were investigated by transfecting cells with various parvin mutants fused to green fluorescent protein (GFP). In general the GFP-fused full-sized parvins co-localized with their endogenous counterparts. Using this method, localization to FAs and fibrillar adhesions was determined to depend on the CH2 domains of both PARVA [2, 4] and PARVB [3]. The CH2 domain alone was sufficient for FA localization, although the other regions appear to increase the intensity of FA localization [2, 4].
A single point mutation (F271D) in the CH2 domain of PARVA, a residue also conserved in PARVB, is sufficient to abolish FA localization [2]. Mutations in the putative paxillin binding site (PBS, amino acids 273–290 of PARVA) also abolish FA localization without affecting ILK binding. Thus, the interaction with ILK is likely necessary but not sufficient for efficient localization of PARVA to FAs. A mutant of ILK (F438A), which does not affect PARVA binding, is unable to localize to FAs, indicating that other interactions are also necessary for FA localization of ILK [19]. Since this site is away from the paxillin binding region, it is more likely that additional proteins, such as Mig-2 or integrins, are also involved in recruitment of the ILK-PARVA complex to the FAs. Since the binding between PARVA and ILK appears stronger than that between ILK and paxillin and the ILK-PARVA complex is formed even in cells without cell-matrix contacts [19], we favor a model whereby PINCH-1, ILK and PARVA form a complex prior to their localization to cell-matrix adhesions. The formation of the PINCH-1-ILK-PARVA complex allows it to be recruited to FAs via interactions with multiple FA proteins such as integrins, paxillin and/or Mig-2 (fig. 2).
Functional role of PARVA phosphorylation
As mentioned earlier, the N-terminal domain of PARVA contains phosphorylation consensus regions for prolinedirected serine/threonine kinase, including ERK and Cdc2. The Cdc2 kinase, in combination with cyclin B1, has been shown to phosphorylate S4, S8, S14 and S19 of PARVA [15], and ERK can also phosphorylate the N-terminus of PARVA [35]. During mitosis, phosphorylation of S4 and S8 by Cdc2 causes gel mobility shift of PARVA. These sites are not present in PARVB.
PARVA phosphorylation increases during cell spreading. Phosphorylation during cell attachment and spreading appears to depend on ERK activity [35]. ERK is recruited to FAs by paxillin during cell spreading. A quintuple mutant of PARVA, in which all four N-terminal serines and T16 were mutated, was not phosphorylated and functioned as dominant negative, inhibiting cell spreading and motility but increasing F-actin formation in human osteosarcoma (U2OS) cells [35]. In kidney podocytes, overexpression of a PARVA mutant, in which the Nterminal proline-directed serine phosphorylation sites are deleted, reduced PARVA complex formation with ILK and inhibited podocyte adhesion, spreading and survival, suggesting an important role of PARVA phosphorylation in regulation of these processes [16].
The interaction of PARVA with TESK1 is also regulated by proline-directed serine phosphorylation of PARVA. Unphosphorylated PARVA binds to TESK1 and inhibits its activity [25], therefore allowing cofilin to reduce actin polymerization and spreading. Upon cell adhesion, ERK is activated to increase the phosphorylation at the N-terminus of PARVA, leading to release of TESK1, phosphorylation of cofilin, F-actin polymerization at the lamellipodia, and increased spreading and motility [25]. Interestingly, ILK was not pulled down by GST-TESK1 [25], indicating that the interactions of PARVA with ILK and TESK1 are mutually exclusive. These results suggest an interesting possibility that phosphorylation of PARVA, which promotes the complex formation of PARVA with ILK and PINCH [16], likely removes the inhibitory effect of PARVA on the interaction with TESK1 and consequently the inhibitory effect on its kinase activity. While the defect in cell spreading caused by mutation of the N-terminus of PARVA was partially corrected by overexpression of TESK1 lacking the PARVA binding site, the defect in cell spreading caused by a PBS mutant of PARVA deficient in paxillin binding was not [25], indicating that PARVA affects cell spreading via multiple signaling pathways.
Regulation of Rac1 activation
The small G-proteins of the Rho family play a critical role in signal transduction pathways connecting integrin signaling and cytoskeletal dynamics [36]. In particular, Rac1 and Cdc42 promote cell spreading and motility, while RhoA promotes formation of actin stress fibers. Rac1 is involved in the formation of wide protrusions, or lamellipodia, associated with early events in cellular attachment and spreading, and in certain types of cellular motility. In contrast, Cdc42 is more involved in the formation of thin protrusion, or filopodia. However, considerable overlap and cross-talking occur between these various Rho family members.
Since both Rac1 and parvins regulate the formation of lamellipodia, it is of great interest to study the interaction between integrins/PINCH/ILK/parvins and Rac1. Depletion of ILK and PINCH-1 significantly (50–55%) reduced adhesion-dependent activation of Rac1 [17]. In striking contrast, depletion of PARVA resulted in a significant increase in Rac1 activity and increased spreading [17]. In contrast, depletion of PARVB in Hela cells did not increase Rac1 activity and cell spreading [17]. In fact, depletion of PARVB in HeLa cells appears to slightly reduce Rac1 activity.
Interestingly, overexpression of the CH1 fragment of PARVB in MDCK cells resulted in Rac1/Cdc42 activation and increased cell spreading and motility with increased formation of lamellipodia and filopodia, an effect blocked by dominant-negative RacN17 and Cdc42N17 [37]. These effects appear to be mediated by interaction of the CH1 domain with aPIX (ARHGEF6), a GEF activator of Cdc42 and Rac1, since an inactive mutant of ARHGEF6 blocked this effect of CH1. Since the interaction of PARVB and aPIX appears to decrease with fulllength PARVB, two possibilities are suggested: (i) ILK complexes with PARVB inhibit interaction with αPIX; and (ii) the C-terminus of PARVB inhibits the interaction with αPIX. Because the CH2 domain of PARVB can be phosphorylated by ILK, it is possible that this induces a conformational change that allows the CH1 domain to bind to αPIX. Alpha-PIX, ILK and PARVB can be found at the leading edge of spreading cells [38]. It is likely that in some cells activation of Rac1 and Cdc42 at the lamellipodia is stimulated by interactions of αPIX with PARVB and regulated by interaction of ILK and PARVB. In addition, PINCH, ILK and parvin proteins may regulate Rac activation through other GEFs such as DOCK180 [39].
Role of parvins in cell spreading and motility
The localization of both PARVA and PARVB at FAs as well as in the leading edge of lamellipodia suggests a role for these proteins in regulating cell adhesion, spreading and motility. Table 2 summarizes studies on the function of parvin, ILK and PINCH proteins in cell adhesion, spreading, motility and actin polymerization. These studies have demonstrated that ILK, PINCH-1, PARVA and PARVB complexes modulate early cellular events triggered by interaction with ECM, including formation and/or turnover of FA, actin dynamics and protrusion of lamellipodia to promote cellular spreading and motility. More specifically, ILK and PINCH-1 are required for all of these processes, since depletion with small interfering RNAs (siRNAs) significantly inhibit them.
The role of the parvins is more complex. Based on their subcellular localization, PARVB appears to play an earlier role in lamellipodia formation. Consistent with an early role in cell adhesion, depletion of PARVB with siRNA caused inhibition of lamellipodia formation [29]. In contrast, depletion of PARVA actually increased cell spreading [22]. As we discussed above, this observation is consistent with the respective roles of PARVA and PARVB in modulating Rac1 activity, which is critical for lamellipodia formation, with PARVA inhibiting and PARVB possibly promoting Rac1 activation. In addition, PARVA suppresses cell spreading through inhibition of TESK1 activity [25]. It will be interesting to test whether PARVB plays an opposite role in regulation of TESK1.
As we have discussed, phosphorylation of PARVA regulates its function and enhances PINCH-ILK-PARVA complex formation. Interestingly, cell adhesion to fibronectin promotes PARVA phosphorylation, which could dissociate the active TESK1 from PARVA and contribute to an increase of actin polymerization and cell spreading. Consistent with these observations, overexpression of unphosphorylatable PARVA mutants (by deletion or mutation of N-terminal sites) acted as dominant-negative inhibitor of cell spreading, presumably by staying locked into an inactive TESK1 complex [25]. Similarly, overexpression of the CH2 domain of PARVA also inhibited cell spreading, despite proper localization to FAs. It is unclear whether CH1 interacting proteins, equivalent to the binding of ARHGEF6 to PARVB, also interact with PARVA and modulate its function at the FAs.
In platelets, an ILK-PARVB complex was rapidly recruited to integrin β3 upon thrombin stimulation, resulting in a transient increase in ILK activity and phosphorylation of PARVB followed by translocation of the ILK-PARVB complex to the cytoskeletal fraction, where it may promote integrin-cytoskeleton interactions [9]. In CHO cells, overexpression of the CH2 domain of PARVB blocked cell spreading. This effect was reversed by cotransfection of ILK with intact kinase activity, suggesting that phosphorylation of PARVB by ILK may play a role in promoting cell spreading [3].
The N-terminus of PARVB CH2 domain (amino acids 249–272) interacts with α-actinin (N-terminal ABD and C-terminal EF-hand domains) only upon engagement of integrins with the ECM, with a peak at 60 min post-adhesion, when cell spreading is most active [29]. The interaction with α-actinin required phosphorylation of the CH2 domain of PARVB by ILK upon cell adhesion. Over-expression of a PARVB mutant containing only amino acids 249–272, which can bind α-actinin but not ILK, disrupted the PARVB-α-actinin interaction and markedly inhibited lamellipodia formation and cell spreading. This effect was also seen with depletion of PARVB by siRNA. These results, together with results from other studies, suggest a model by which a PINCH-ILK-PARVB complex forms at integrin attachment sites in very early membrane blebs, where ILK kinase activity is induced, resulting in phosphorylation of the PARVB CH2 domain. PARVB then recruits α-actinin (or stabilizes the integrin-α-actinin complex) to the leading edge of the lamellipodia. PINCH, ILK, PARVB and α-actinin can promote lamellipodia extension by attracting actin polymerization complexes, including proteins of the Nck and ARP2/3 (through Rac/ Cdc42/WASP) families [21, 40–43].
Role of parvins on Akt activation and cell survival
PARVA plays an important role in protection of cells from apoptosis [44]. By contrast, depletion of PARVB in HeLa cells does not induce apoptosis, indicating it is not essential for cell survival [17]. PARVA protects cells from apoptosis by facilitating the activation of Akt (protein kinase B), a key survival signaling intermediate. In response to cell-matrix adhesion and soluble survival factors such as IGF-1, inactive Akt is translocated to plasma membrane, where it is phosphorylated on T308 and S473 and consequently fully activated. Loss of PARVA impairs the membrane translocation of Akt [44]. Furthermore, expression of a constitutively membrane-bound (myristoylated) Akt (Myr-Akt) rescued the defects in the activating phosphorylation of Akt and cell survival induced by the loss of PARVA. Thus, PARVA protects cells from apoptosis via facilitating the activating phosphorylation of Akt [44].
ILK depletion also affects Akt phosphorylation and activation in some cell types, particularly transformed cells (reviewed in [13, 45]). ILK kinase activity is transiently increased during cell attachment and enriched in the cytoskeletal fraction [18]. PARVA-depleted cells show much lower ILK kinase activity [18], indicating that the formation of the PARVA-ILK complex also plays a role for the activation of ILK. Mutations in ILK that abolish the ILK-PARVA interaction also inhibit the ability of ILK to promote Akt S473 phosphorylation [18, 46]. In cells overexpressing PTEN, ILK kinase activity and PARVA interaction are inhibited. Transfection of PARVA restores ILK activity, as demonstrated by increased ILK kinase activity, GSK3β S9 phosphorylation, Akt S473 phosphorylation and β-catenin/TCF/LEF transcriptional activity [18]. Although ILK and PINCH-1 likely work in concert with PARVA in regulation of Akt phosphorylation, Akt is not the only downstream target in ILK and PINCH-1-mediated cell survival signaling [22].
Role of parvins in striated muscle
In C. elegans, the integrin-dependent pathways involved in myofibrillogenesis have been clarified by forward genetic screens. Deposition of the ECM protein perlecan/UNC-52 initiates outside-in signaling transduced by the integrin receptor, PAT-3, which then recruits two complexes: (i) vinculin/DEB1, and (ii) ILK/PAT-4 + Mig-2/UNC-112 [5]. Both of these complexes are required for the formation of rudimentary FAs, which will become the initial sites of myofibril formation (the dense bodies) and subsequently of muscle attachment to the body wall. Maturation of dense bodies involves recruitment of PARVA/PAT-6 and PINCH-1/UNC-97, both of which bind to ILK/PAT-4, as determined by yeast three-hybrid assay [5]. In addition to this PINCH-ILK-parvin complex, vinculin and talin are required for initiation of myofibril formation by attracting sarcomeric actin filaments [5]. Alpha-actinin seems to be added to functional dense bodies at a later stage after larval hatching. Recruitment of myosin filaments does not require vinculin; instead UNC-89 is required at the M-line [5]. Interestingly, UNC-89 is the ortologue of an alternatively spliced isoform of obscurin containing several immunoglobulin domains and a Rho guanidine nucleotide exchange factor (GEF) domain. Obscurin and titin co-localize during myofibrillogenesis in cardiac myocytes [47], and the obscurin-RhoGEF isoform is upregulated during cardiac hypertrophy [48–50]. Since obscurin interacts with titin at the Z-line [48] and PARVA-ILK complexes are localized in the costameres overlapping the Z-line of cardiac myocytes and play a role in myocyte hypertrophy [51], these observations suggest that modulation of obscurin activity and/or localization by ILK-PARVA complexes may play a role in sarcomere assembly during cardiac hypertrophy. In concordance with the studies in mammalian cells, the CH2 domain of PARVA/PAT6 was necessary and sufficient for binding to ILK and for efficient localization to dense bodies. The CH1 domain was dispensable for proper localization, but it was required for downstream events leading to myofibrillogenesis.
Recent studies have demonstrated the presence of a PINCH-1-ILK-PARVA complex in neonatal cardiac myocytes, and its recruitment to costameres associated with the Z-disks of newly formed sarcomeres in response to hypertrophic stimulation with fibronectin and phenylephrine [51]. A dominant-negative mutant of ILK lacking PINCH-1 binding (ILK-C) removed ILK and PARVA from costamere locations and induced mild hypertrophy, while promoting apoptosis. These observations suggest a role for PARVA in promoting cell survival and inhibiting hypertrophy in the cardiac myocyte. Since PARVA inhibits Rac1 activity and this small G-protein plays a critical role in cardiac hypertrophy [52], we are currently investigating the possibility that PARVA and other ILK complexes regulate the level of Rac1 activity in cardiac myocytes.
PARVB and ILK co-localized at regions of the sarcolemma corresponding to the Z-disk in skeletal muscle (costameres) [3]. In another study, PARVB co-localized and interacted with the sarcolemmal protein, dysferlin, in human skeletal muscle [53]. The sarcolemmal localization of PARVB was reduced in muscle dystrophy with dysferlin mutations and may indicate a role for PARVB in membrane integrity. Taken together, these results suggest that parvin proteins may play both physical and signaling roles in muscle cells.
Role of parvins in pathological processes
Given the importance of the parvins in regulating actin cytoskeleton and survival signaling, they are probably involved in a number of pathological processes. In particular, recent studies have implicated parvins in two human diseases, namely breast cancer and diabetic nephropathy.
Breast cancer
Anchorage-independent growth and resistant to apoptosis induced by loss of integrin-ECM contact (anoikis) is a hallmark of cancer cells. Moreover, changes in cell-ECM interaction can lead to increased motility characteristic of metastatic cells. Overexpression of ILK with hyperactive kinase function has been shown to occur in several transformed epithelial cells [54] and inhibition of ILK kinase or expression of ILK dominant-negative mutants can reverse several features of the transformed phenotype [18, 54]. The respective roles of PARVA and PARVB in this oncogenic role of ILK have not been fully characterized. PARVA has been shown to promote anti-apoptotic signaling from activated ILK [18, 44]. In contrast, in breast cancer PARVB appears to be an inhibitor of ILK kinase [6]. In one recent study, PARVB mRNA and protein levels were found to be markedly downregulated in a number of advanced tumors, together with increases in ILK protein level and kinase activity [6]. Low PARVB levels correlated with low adhesion to collagen and restoring the levels of PARVB resulted in increased adhesion, and reversal of anchorage-independent growth coincident with decreased ILK activity. In addition, overexpression of PARVB increases apoptosis [17]. The activating phosphorylation of Akt was reduced by increased levels of PARVB [6], which may translate in increased susceptibility of the cancer cells to apoptosis. Another target of ILK is GSK3β, which can regulate nuclear accumulation of β-catenin, frequently observed in transformed cells. Increased ILK activity leads to nuclear accumulation of β-catenin and upregulation of TCF/LEF targets, whereas restoring PARVB levels in cancer cells appears to downregulate nuclear β-catenin [6]. This tumor-suppressive role of PARVB is consistent with deletions in the PARVB locus (22q13.21) observed in some colon and breast cancers [55]. The PARVG gene was suspected to have a similar tumor suppressive role in a locus 12 Kb away (22q13.31), but no mutations were identified in colon and breast tumors [56].
Kidney disease
ILK, PARVA and PINCH-1 are expressed in glomerular mesangial cells and podocytes [57]. By mRNA differential display analysis, ILK was identified as a candidate gene in Finnish type congenital nephritic proteinuria [58]. ILK expression was found to be increased in glomeruli [57], which could contribute to the increased matrix deposition in glomerulosclerosis in human patients.
ILK is involved in mesangial cell matrix deposition in response to hyperglycemia [57]. In mesangial cells, the PINCH-1-ILK-PARVA complex locates to FAs and fibrillar adhesions where it promotes fibronectin matrix deposition [33]. Disruption of the PINCH-1-ILK-PARVA complex with ILK-C or ILK-N dominant-negative mutants resulted in decreased cell proliferation, fibronectin synthesis and matrix deposition [33].
The levels of PINCH-1, ILK and PARVA increase with podocyte differentiation, and the PINCH-1-ILK-PARVA complex plays a critical role in podocyte adhesion, architecture and survival [16]. In these cells, interference with the formation of the PINCH-1-ILK-PARVA complex by dominant-negative ILK and PARVA constructs reduced podocyte adhesion, spreading and survival. ILK also promotes upregulation of matrix metalloproteinase-9 (MMP-9) mRNA levels and β-catenin translocation during podocyte stress [59–61]. In addition to its role in the glomerulus, ILK complexes play an important role in tubular cell epithelial to mesenchymal transition and interstitial fibrosis in response to TGF-β, tubular obstruction or hyperglycemia [62]. A dominant-negative mutant of ILK largely reversed these effects.
Given the important role of the ILK-PINCH1-PARVA complex in renal physiology and pathology, this complex is a potential target for therapeutic intervention, provided that beneficial effects, such as podocyte adhesion and survival, are not significantly inhibited.
Conclusions and Future Studies
From the studies discussed in this review, it is apparent that PARVA and PARVB play critical roles in several cell functions, regulating cytoskeletal activity in connection with the ECM, and that these functions are mediated by their ability to form complexes with key proteins at the different subcellular locations, such as FAs, lamellipodia, fibrillar adhesions and possibly nuclei. In particular, the formation and regulation of complexes involving parvin, ILK and PINCH proteins as well as interactions with other ancillary proteins, such as Mig-2 [63], paxillin, ARHGEF6, α-actinin and TESK1, together with differential phosphorylation by Cdc2, ERK and ILK, mediate the various effects of PARVA and PARVB. Clearly, despite their high homology, different members of the parvin family have distinct functions imparted by their ability to form alternative complexes and differential regulation. Table 3 summarizes some of the features distinguishing the two parvins.
Despite considerable progress, much remains to be investigated to clarify the roles of parvins in cell physiology and pathological processes. For example, PARVG appears similar to PARVA and PARVB, but as we have learned, similar domains could mediate quite different effects. Clarifying the role of PARVG, particularly in hematological cells and pathologies, will be an interesting area of future research. Further investigation on the various PINCH-ILK-parvin complexes and their binding proteins in different cell types under different stimulatory events should help us to elucidate the role of each complex in cell physiology. Of particular interest is the elucidation of the mechanisms by which PARVA and PARVB regulate the activity of Rho family GTPases in the various cell types. Searching for additional proteins interacting with the CH domains, in particular the CH1 domain of PARVA and the CH2 of PARVB, should reveal additional details on how parvin proteins regulate cell spreading, motility and survival.
Undoubtedly, this field will continue to progress at a rapid pace. Further interactions and cross-talking pathways involving parvins and their partners will be uncovered. Ultimately, only a comprehensive systems biology approach well-grounded on experimental observation and powerful computational resources will be able to model and make reliable predictions about the functioning of this important complex network in transducing signaling between the ECM and the cell.
References
Nikolopoulos S. N. and Turner C. E. (2000) Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell. Biol. 151: 1435–1448
Tu Y., Huang Y., Zhang Y., Hua Y. and Wu C. (2001) A new focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J. Cell. Biol. 153: 585–598
Yamaji S., Suzuki A., Sugiyama Y., Koide Y., Yoshida M., Kanamori H. et al. (2001) A novel integrin-linked kinase-binding protein, affixin, is involved in the early stage of cell-substrate interaction. J. Cell. Biol. 153: 1251–1264
Olski T. M., Noegel A. A. and Korenbaum E. (2001) Parvin, a 42 kDa focal adhesion protein, related to the alpha-actinin superfamily. J. Cell. Sci. 114: 525–538
Lin X., Qadota H., Moerman D. G. and Williams B. D. (2003) C. elegans PAT-6/actopaxin plays a critical role in the assembly of integrin adhesion complexes in vivo. Curr. Biol. 13: 922–932
Mongroo P. S., Johnstone C. N., Naruszewicz I., Leung-Hagesteijn C., Sung R. K., Carnio L. et al. (2004) Beta-parvin inhibits integrin-linked kinase signaling and is downregulated in breast cancer. Oncogene 23: 8959–8970
Korenbaum E., Olski T. M. and Noegel A. A. (2001) Genomic organization and expression profile of the parvin family of focal adhesion proteins in mice and humans. Gene 279: 69–79
Gimona M., Djinovic-Carugo K., Kranewitter W. J. and Winder S. J. (2002) Functional plasticity of CH domains. FEBS Lett. 513: 98–106
Yamaji S., Suzuki A., Kanamori H., Mishima W., Takabayashi M., Fujimaki K. et al. (2002) Possible role of ILK-affixin complex in integrin-cytoskeleton linkage during platelet aggregation. Biochem. Biophys. Res. Commun. 297: 1324–1331
Hannigan G. E., Leung-Hagesteijn C., Fitz-Gibbon L., Coppolino M. G., Radeva G., Filmus J. et al. (1996) Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379: 91–96
Wu C. and Dedhar S. (2001) Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell. Biol. 155: 505–510
Grashoff C., Thievessen I., Lorenz K., Ussar S. and Fassler R. (2004) Integrin-linked kinase: integrin’s mysterious partner. Curr. Opin. Cell Biol. 16: 565–571
Hannigan G., Troussard A. A. and Dedhar S. (2005) Integrinlinked kinase: a cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 5: 51–63
Nikolopoulos S. N. and Turner C. E. (2002) Molecular dissection of actopaxin-integrin-linked kinase-Paxillin interactions and their role in subcellular localization. J. Biol. Chem. 277: 1568–1575
Curtis M., Nikolopoulos S. N. and Turner C. E. (2002) Actopaxin is phosphorylated during mitosis and is a substrate for cyclin B1/cdc2 kinase. Biochem. J. 363: 233–242
Yang Y., Guo L., Blattner S. M., Mundel P., Kretzler M. and Wu C. (2005) Formation and phosphorylation of the PINCH-1-in-tegrin linked kinase-{alpha}-parvin complex are important for regulation of renal glomerular podocyte adhesion, architecture and survival. J. Am. Soc. Nephrol. 16: 1966–1976
Zhang Y., Chen K., Tu Y. and Wu C. (2004) Distinct roles of two structurally closely related focal adhesion proteins, alpha-parvins and beta-parvins, in regulation of cell morphology and survival. J. Biol. Chem. 279: 41695–41705
Attwell S., Mills J., Troussard A., Wu C. and Dedhar S. (2003) Integration of cell attachment, cytoskeletal localization and signaling by integrin-linked kinase (ILK), CH-ILKBP and the tumor suppressor PTEN. Mol. Biol. Cell 14: 4813–4825
Zhang Y., Chen K., Tu Y., Velyvis A., Yang Y., Qin J. et al. (2002) Assembly of the PINCH-ILK-CH-ILKBP complex precedes and is essential for localization of each component to cell-matrix adhesion sites. J. Cell. Sci. 115: 4777–4786
Tu Y., Li F., Goicoechea S. and Wu C. (1999) The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol. 19: 2425–2434
Wu C. (1999) Integrin-linked kinase and PINCH: partners in regulation of cell-extracellular matrix interaction and signal transduction. J. Cell. Sci. 112(Pt 24): 4485–4489
Fukuda T., Chen K., Shi X. and Wu C. (2003) PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility and survival. J. Biol. Chem. 278: 51324–51333
Turner C. E. (2000) Paxillin interactions. J. Cell. Sci. 113Pt 23: 4139–4140
Turner C. E. (2000) Paxillin and focal adhesion signalling. Nat. Cell Biol. 2: E231–236
LaLonde D. P., Brown M. C., Bouverat B. P. and Turner C. E. (2005) Actopaxin interacts with TESK1 to regulate cell spreading on fibronectin. J. Biol. Chem. 280: 21680–21688
Tsumura Y., Toshima J., Leeksma O. C., Ohashi K. and Mizuno K. (2005) Sprouty-4 negatively regulates cell spreading by inhibiting the kinase activity of testicular protein kinase. Biochem. J. 387: 627–637
Toshima J. Y., Toshima J., Watanabe T. and Mizuno K. (2001) Binding of 14-3-3beta regulates the kinase activity and subcellular localization of testicular protein kinase 1. J. Biol. Chem. 276: 43471–43481
Leeksma O. C., Van Achterberg T. A., Tsumura Y., Toshima J., Eldering E., Kroes W. G. et al. (2002) Human sprouty 4, a new ras antagonist on 5q31, interacts with the dual specificity kinase TESK1. Eur. J. Biochem. 269: 2546–2556
Yamaji S., Suzuki A., Kanamori H., Mishima W., Yoshimi R., Takasaki H. et al. (2004) Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J. Cell. Biol. 165: 539–551
Rosenberger G., Jantke I., Gal A. and Kutsche K. (2003) Interaction of alphaPIX (ARHGEF6) with beta-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum. Mol. Genet. 12: 155–167
Zaidel-Bar R., Cohen M., Addadi L. and Geiger B. (2004) Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 32: 416–420
Geiger B., Bershadsky A., Pankov R. and Yamada K. M. (2001) Transmembrane crosstalk between the extracellular matrix — cytoskeleton crosstalk. Nat. Rev. Mol. Cell. Biol. 2: 793–805
Guo L. and Wu C. (2002) Regulation of fibronectin matrix deposition and cell proliferation by the PINCH-ILK-CH-ILKBP complex. FASEB J. 16: 1298–1300
Nikolopoulos S. N. and Turner C. E. (2001) Integrin-linked kinase (ILK) binding to paxillin LD1 motif regulates ILK localization to focal adhesions. J. Biol. Chem. 276: 23499–23505
Clarke D. M., Brown M. C., LaLonde D. P. and Turner C. E. (2004) Phosphorylation of actopaxin regulates cell spreading and migration. J. Cell. Biol. 166: 901–912
Jaffe A. B. and Hall A. (2005) RHO GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21: 247–269
Mishima W., Suzuki A., Yamaji S., Yoshimi R., Ueda A., Kaneko T. et al. (2004) The first CH domain of affixin activates Cdc42 and Rac1 through alphaPIX, a Cdc42/Rac1-specific guanine nucleotide exchanging factor. Genes Cells 9: 193–204
Rosenberger G., Gal A. and Kutsche K. (2005) AlphaPIX associates with calpain 4, the small subunit of calpain, and has a dual role in integrin-mediated cell spreading. J. Biol. Chem. 280: 6879–6889
Tu Y., Kucik D. F. and Wu C. (2001) Identification and kinetic analysis of the interaction between Nck-2 and DOCK180. FEBS Lett. 491: 193–199
Tu Y., Li F. and Wu C. (1998) Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase-signaling pathways. Mol. Biol. Cell. 9: 3367–3382
DeMali K. A., Wennerberg K. and Burridge K. (2003) Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol. 15: 572–582
Vaynberg J., Fukuda T., Chen K., Vinogradova O., Velyvis A., Tu Y. et al. (2005) Structure of an ultraweak protein-protein complex and its crucial role in regulation of cell morphology and motility. Mol. Cell 17: 513–523
Xu Z., Fukuda T., Li Y., Zha X., Qin J. and Wu C. (2005) Molecular dissection of PINCH-1 reveals a mechanism of coupling and uncoupling of cell shape modulation and survival. J. Biol. Chem. 280: 27631–27637
Fukuda T., Guo L., Shi X. and Wu C. (2003) CH-ILKBP regulates cell survival by facilitating the membrane translocation of protein kinase B/Akt. J. Cell. Biol. 160: 1001–1008
Wu C. (2004) The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim. Biophys. Acta 1692: 55–62
Persad S., Attwell S., Gray V., Mawji N., Deng J. T., Leung D. et al. (2001) Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 276: 27462–27469
Sepulveda J., Gkretsi V. and Wu C. (2005) Assembly and signaling of adhesion complexes. Curr. Top. Dev. Biol. 68: 183–225
Young P., Ehler E. and Gautel M. (2001) Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J. Cell Biol. 154: 123–136
Borisov A. B., Raeker M. O., Kontrogianni-Konstantopoulos A., Yang K., Kurnit D. M., Bloch R. J. et al. (2003) Rapid response of cardiac obscurin gene cluster to aortic stenosis: differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem. Biophys. Res. Commun. 310: 910–918
Borisov A. B., Kontrogianni-Konstantopoulos A., Bloch R. J., Westfall M. V. and Russell M. W. (2004) Dynamics of obscurin localization during differentiation and remodeling of cardiac myocytes: obscurin as an integrator of myofibrillar structure. J. Histochem. Cytochem. 52: 1117–1127
Chen H., Huang X. N., Yan W., Chen K., Guo L., Tummalapali L. et al. (2005) Role of the integrin-linked kinase/PINCH1/alpha-parvin complex in cardiac myocyte hypertrophy. Lab. Invest. Sep. 10 [Epub ahead of print]
Pracyk J. B., Tanaka K., Hegland D. D., Kim K. S., Sethi R., Rovira I. I. et al. (1998) A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J. Clin. Invest. 102: 929–937
Matsuda C., Kameyama K., Tagawa K., Ogawa M., Suzuki A., Yamaji S. et al. (2005) Dysferlin interacts with affixin (betaparvin) at the sarcolemma. J. Neuropathol. Exp. Neurol. 64: 334–340
Persad S. and Dedhar S. (2003) The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 22: 375–384
Castells A., Gusella J. F., Ramesh V. and Rustgi A. K. (2000) A region of deletion on chromosome 22q13 is common to human breast and colorectal cancers. Cancer Res. 60: 2836–2839
Castellvi-Bel S., Castells A., Johnstone C. N., Pinol V., Pellise M., Elizalde J. I. et al. (2003) Evaluation of PARVG located on 22q13 as a candidate tumor suppressor gene for colorectal and breast cancer. Cancer Genet. Cytogenet. 144: 80–82
Guo L., Sanders P. W., Woods A. and Wu C. (2001) The distribution and regulation of integrin-linked kinase in normal and diabetic kidneys. Am. J. Pathol. 159: 1735–1742
Kretzler M., Teixeira V. P., Unschuld P. G., Cohen C. D., Wanke R., Edenhofer I. et al. (2001) Integrin-linked kinase as a candidate downstream effector in proteinuria. FASEB J. 15: 1843–1845
Troussard A. A., Costello P., Yoganathan T. N., Kumagai S., Roskelley C. D. and Dedhar S. (2000) The integrin linked kinase (ILK) induces an invasive phenotype via AP-1 transcription factor-dependent upregulation of matrix metalloproteinase 9 (MMP-9). Oncogene 19: 5444–5452
von Luttichau I., Djafarzadeh R., Henger A., Cohen C. D., Mojaat A., Jochum M. et al. (2002) Identification of a signal transduction pathway that regulates MMP-9 mRNA expression in glomerular injury. Biol. Chem. 383: 1271–1275
Teixeira Vde P., Blattner S. M., Li M., Anders H. J., Cohen C. D., Edenhofer I. et al. (2005) Functional consequences of integrin-linked kinase activation in podocyte damage. Kidney Int. 67: 514–523
Li Y., Yang J., Dai C., Wu C. and Liu Y. (2003) Role for integrinlinked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J. Clin. Invest. 112: 503–516
Tu Y., Wu S., Shi X., Chen K. and Wu C. (2003) Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 113: 37–47
Acknowledgement
This work was supported by NIH grants GM65188 and DK54639 to C.W. and HL068714 to J.S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 5 August 2005; received after revision 5 September 2005; accepted 22 September 2005
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sepulveda, J.L., Wu, C. The parvins. Cell. Mol. Life Sci. 63, 25–35 (2006). https://doi.org/10.1007/s00018-005-5355-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-005-5355-1