
Abstract
Background
Common and rare variants of guanosine triphosphate cyclohydrolase 1 (GCH1) gene may play important roles in Parkinson’s disease (PD). However, there is a lack of comprehensive analysis of GCH1 genotypes, especially in non-coding regions. The aim of this study was to explore the genetic characteristics of GCH1, including rare and common variants in coding and non-coding regions, in a large population of PD patients in Chinese mainland, as well as the phenotypic characteristics of GCH1 variant carriers.
Methods
In the first cohort of this case-control study, we performed whole-exome sequencing in 1555 patients with early-onset or familial PD and 2234 healthy controls; then in the second cohort, whole-genome sequencing was performed in sporadic late-onset PD samples (1962 patients), as well as 1279 controls. Variants at target GCH1 regions were extracted, and then genetic and detailed phenotypic data were analyzed using regression models and the sequence kernel association test. We also performed a meta-analysis to correlate deleterious GCH1 variants with age at onset (AAO) in PD patients.
Results
For coding variants, we identified a significant burden of GCH1 deleterious variants in early-onset or familial PD cases compared to controls (1.2% vs 0.1%, P < 0.0001). In the analysis of possible regulatory variants in GCH1 non-coding regions, rs12323905 (P = 0.001, odds ratio = 1.19, 95%CI 1.07–1.32) was significantly associated with PD, and variant sets in untranslated regions and intron regions, GCH1 brain-specific expression quantitative trait loci, and two possible promoter/enhancer (GH14J054857 and GH14J054880) were suggestively associated with PD. Genotype-phenotype correlation analysis revealed that the carriers of GCH1 deleterious variants manifested younger AAO (P < 0.0001), and had milder motor symptoms, milder fatigue symptoms and more autonomic nervous dysfunctions. Meta-analysis of six studies demonstrated 6.4-year earlier onset in GCH1 deleterious variant carriers (P = 0.0009).
Conclusions
The results highlight the importance of deleterious variants and non-coding variants of GCH1 in PD in Chinese mainland and suggest that GCH1 mutation can influence the PD phenotype, which may help design experimental studies to elucidate the mechanisms of GCH1 in the pathogenesis of PD.
Similar content being viewed by others


Background
Parkinson’s disease (PD) is the second most common neurodegenerative disease characterized clinically by bradykinesia, resting tremor, rigidity, and postural instability. The interplay among aging, genetics and environmental factors plays important roles in PD pathogenesis [1, 2]. The core biological feature of PD is nigrostriatal dopamine deficiency, which is shared by dopa-responsive dystonia (DRD), a disorder characterized by childhood- or adolescent-onset dystonia and dramatic response to low doses of L-dopa without degeneration of the nigral neurons [3, 4]. A significant proportion of early-onset PD patients manifest with dystonia [5]; moreover, co-occurrence of DRD and parkinsonism has been reported in families with mutations of guanosine triphosphate cyclohydrolase 1 (GCH1) gene [6], which is a well-established disease-causing gene for DRD. GCH1 plays a pivotal role in dopamine biosynthesis [7], indicating that it may also participate in the pathogenesis of PD. Exome-wide analysis has revealed increased burden of predicted deleterious GCH1 mutations in PD patients [8,9,10], and genome-wide association studies (GWAS) have identified associations of polymorphisms in GCH1 locus with PD [11, 12]. However, conflicting results have been reported among different populations [13, 14], indicating the genetic heterogeneity among populations. Thus, it is important to explore whether rare and common GCH1 variants play a role in PD patients in Chinese mainland. Furthermore, there is still a lack of studies on GCH1 variants in regulatory regions such as promoter, enhancer, and expression quantitative trait loci (eQTLs). In addition, mutation and polymorphism in the regulatory regions may influence the transcriptional activity by affecting the binding and regulatory ability of transcription factors [15]. Therefore, it is necessary to explore the relationship between variants in GCH1 regulatory elements and PD.
In addition to the effect on the risk of PD, genetic variants may also play roles in specific clinical phenotypes [16, 17]. With the development of precision medicine, understanding the differences in clinical phenotypes of varied genetic forms of PD may help clinicians in symptomatic treatment and consultation. Previous studies [8,9,10] have only described specific symptoms of GCH1 variant-carriers, and there is a lack of systematical analysis of clinical manifestations of GCH1 variant-carriers. Studies have shown that monogenic genes in PD, such as PRKN and PINK1 [18], and rare damaging variants in PD risk gene such as GBA [19], are associated with an earlier age at onset (AAO), which suggested the importance of genetic variants for AAO. Therefore, it is necessary to study the effect of GCH1 variants on clinical manifestation of PD in our study, especially the AAO.
In the present study, we first analyzed the burden of GCH1 variants in coding regions in PD patients using whole-exome sequencing (WES), and then examined rare and common variants in the reported regulatory elements of GCH1 including promoter, enhancer, untranslated regions (UTRs)/introns and eQTLs, using whole-genome sequencing (WGS). Third, we analyzed the clinical manifestations of PD patients with GCH1 rare or common variants, especially in terms of AAO.
Materials and methods
Participants
PD patients of Chinese origin who visited the Department of Neurology, Xiangya Hospital between October 2006 and January 2019 and at other cooperating centers of Parkinson’s Disease & Movement Disorders Multicenter Database and Collaborative Network in China (PD-MDCNC, http://pd-mdcnc.com:3111/) were recruited in the study. They were diagnosed by movement disorder specialists according to the UK brain bank criteria [20] or Movement Disorders Society (MDS) clinical diagnostic criteria [21]. Control participants without any neurological diseases were recruited from community or were the spouse of the recruited patients. The patients and controls were divided into two cohorts, cohort WES and cohort WGS.
The cohort WES included PD patients with AAO no more than 50 years or with a family history of PD, and control participants, who were assessed using WES. One hundred and twenty-one patients with pathogenic/likely pathogenic variants of high- or very high-confidence PD disease-causing genes [22] were excluded from the cohort and the related study was accepted and in press. A large sample of sporadic late-onset (AAO > 50) PD patients, and the age/sex-matched healthy controls were included in the cohort WGS.
All participants provided written informed consent for participation in the genetic research, which was approved by relevant oversight committees and institutional review boards. Genomic DNA was prepared from peripheral blood leukocytes according to standard procedures. Of note, 13.8% (973/7030) of the participants overlapped with those in our previous study [10] of rare GCH1 variants in PD patients analyzed by molecular inversion probe technique, including 891 cases and 82 controls.
A comprehensive dataset of basic demographic data including age, sex, family history, disease duration and clinical features including motor and non-motor manifestations, was collected from the recruited PD patients, and saved into the PD-MDCNC database.
Genotyping and quality control
WES and WGS were performed according to previous descriptions [23, 24]. Burrows-Wheeler Aligner-MEM algorithm [25], Picard (http://broadinstitute.github.io/picard/), and Genome Analysis Toolkit [26] were used to generate high-quality variants. ANNOVAR [27] and VarCards [28] were used to annotate the variants with gene regions (RefSeq, hg19), amino acid alterations, functional effects, and allele frequencies in East Asian population (gnomAD database, ExAC database). And ReVe [29] was used for in silico pathogenicity prediction (threshold, 0.7).
For individual and variant quality control, the PLINK software v1.90 was used [30]. Individuals were excluded if they showed ambiguous sex (conflicting sex assignment in PLINK), low genotype call rates (missing rate > 10%), deviating heterozygosity/genotype calls (± 3 standard deviations [SDs]), or cryptic relatedness (identity by descent > 0.15). Variant quality control was accomplished by removal of low-quality genotypes (Phred-scaled genotype quality score below 30, allele depth [AD] below 10, and reads depth [DP] below 30 for WES data; Phred-scaled genotype quality score below 15, AD below 2, and DP below 5 for WGS data) and variants with low call rates (missing rate > 10%) or departure from Hardy-Weinberg equilibrium (P < 0.0001). Principal component analysis for population stratification was conducted using independent high-quality variants and main principal component variables for each sample were obtained. Outliers (suggesting non-Chinese ancestry) were excluded from further analysis.
Selection of GCH1 variants
The GCH1 transcript region including UTRs, exons and introns (NM_000161 at chr14:55,308,724-55,369,542; hg19) [31], regulatory region from Genehancer [32], brain-specific eQTLs from Genotype-Tissue Expression (GTEx) project [33], and significant GWAS signals [12] were considered in the study (Table S1), and the corresponding quality parameters of the targeted GCH1 regions are presented in Table S2. The variants from target GCH1 regions were extracted and divided into coding variants or non-coding variants as per their position in the genome, and common or rare variants on the basis of the minor allele frequencies (MAF) in covered samples of cohorts WES and WGS at a threshold of 0.01 (MAF < 0.01 into rare variants and others into common variants). The coding variants were further categorized into missense, synonymous, loss-of-function (stop gain/loss, frameshift, and splicing mutations falling within two base pairs of exon-intron junctions), protein-altering (missense and loss-of-function), and deleterious (predicted to be damaging by ReVe or previously reported to be associated with PD or DRD). Specifically, the Gene4PD database (http://www.genemed.tech/gene4pd/) developed by our group, the MDSgene database (https://www.mdsgene.org/), and PubMed were searched to determine if these variants had been reported [8, 10, 34,35,36,37,38] to be associated with PD or DRD. The non-coding variants were assorted into UTRs/introns, eQTLs, GWAS signals, and regulatory elements according to the genomic location and functional property.
Meta-analysis
Literature was searched in electronic databases including PubMed, Embase and Cochrane Library using a combination of following keywords: “GCH1”, “guanosine triphosphate cyclohydrolase 1”, “GTP cyclohydrolase 1”, “Parkinson disease”, “Parkinson’s disease”, and “Parkinsonism”. The last search was updated on April 1, 2020. Two researchers performed the search independently and in case of a disagreement, a third researcher was consulted to arrive at a consensus.
Eligible studies were included according to the following criteria: (1) should be an observational study, such as using a case-control or cohort design; (2) all PD patients should be diagnosed according to the UK brain bank criteria [20] or MDS clinical diagnostic criteria for PD [21]; and (3) clearly reporting results of deleterious GCH1 variants and corresponding AAO data. Moreover, the following studies were excluded: (1) case reports, editorials, reviews or functional research; (2) duplicate studies (when multiple studies employed the same participants, the latest or most complete report was included); and (3) studies with incomplete data, which did not specify the AAO among carriers of GCH1 deleterious variants.
Data were extracted for meta-analysis by two researchers independently, including first author’s name, year of publications, ethnicity and country of participants, gene and variants, numbers of PD patients with and without GCH1 deleterious variants, and their corresponding AAO information. Any disagreement was resolved by the senior authors. The quality of the included studies was evaluated by the Newcastle-Ottawa Scale [39], which came out to be approximately good for the present study.
Statistical analysis
For common variant association analysis, PLINK [30] was used to perform logistic regression with age, sex and first five principal components for population stratification as covariates. To adjust for multiple comparisons, Bonferroni-corrected significance threshold was used to determine the statistical difference. We applied spectral decomposition adjustment using the SNPSpD method (https://gump.qimr.edu.au/general/daleN/SNPSpD) to determine the number of independent statistical tests in over 200 common variants. The source of each common variant is shown in Table S3. The estimated number of independent tests was 19, corresponding to a Bonferroni-corrected significance threshold of P < 0.003.
For variant set analysis, the sequence kernel association test (SKAT) implemented by the SKAT R package was used. The optimized SKAT (SKAT-O) was used to analyze the rare variant association in a specified region and combined SKAT (SKAT-C) was used to evaluate the cumulative effect of all variants including both rare and common variants on disease risk [40, 41]. Covariates were included to adjust the analyses for sex, age and first five principal components of ancestry. The estimated number of independent tests was 11, corresponding to a Bonferroni-corrected significance threshold of P < 0.0045. We considered those results with a P-value < 0.05, but not surviving the Bonferroni correction, as ‘suggestive’.
For genotype-phenotype correlation assessment, linear and logistic regression analyses were performed using PLINK, with adjustment for age at entry, disease duration, and sex. The estimated number of independent tests was 22, corresponding to a Bonferroni-corrected significance threshold of P < 0.0023.
Meta-analysis of the association of deleterious variants of GCH1 with AAO of PD patients was carried out, which included our own data from this study. To assess the importance of GCH1 deleterious variants on the AAO of PD patients, continuous data were expressed as pooled mean difference with 95% confidence intervals (CI). The heterogeneity across studies was evaluated by Higgin’s I2 index. If the heterogeneity (P > 0.10, I2 < 50%) was not significant, the fixed effect model was adopted, otherwise the random effect model was used. The reporting biases were assessed by funnel-plot analysis. All analyses were done using the Review Manager software package v.5.3 (The Cochrane Collaboration, Oxford, England).
Results
Cohort description
The WES cohort included 1555 PD patients with AAO ≤ 50 years or with a family history, and 2234 controls without any neurological disease. The PD patients had a mean age of 52.3 ± 8.9 years and a mean AAO of 46.0 ± 8.3 years; 54.3% (845/1555) of them were male and 23.0% (358/1555) reported a positive family history. The mean age of control participants was 42.8 ± 8.7 years and 60.2% (1345/2234) were male. The average sequencing depth in the WES cohort was 123-fold and 99.32% of targeted regions achieved a minimum of 10 coverage.
The WGS cohort consisted of 1962 PD patients with sporadic late-onset PD (AAO > 50 years) and 1279 age/sex-matched controls. The PD cases were recruited at a mean age of 66.8 ± 7.1 years and had a mean AAO of 61.9 ± 6.9 years, and 50.2% (984/1962) were male. Control participants had an average age of 59.3 ± 7.1 years and 48.0% (613/1279) were male. The average sequencing depth in the WGS cohort was 12-fold and 95.5% of genome regions were covered at least 5-fold.
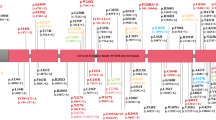
The workflow of the study is presented in Fig. 1. The detailed information about the target variants and regions related to GCH1 included in the study are shown in Table S1. The baseline characteristics of the demographic data, the motor and non-motor symptoms of patients from cohort WES and cohort WGS are presented in Table S4.

Workflow of this study
Coding variants
Both cohorts WES and WGS were subjected to coding variant analysis, which included six exons of the coding variants. Of note, all the identified coding variants were rare (Table S5).
In the WES cohort, a significant association of GCH1 coding variants with PD was found (1.6% vs 0.5%, SKAT-O, P = 0.0001). Further stratification by variant properties showed that the association mainly involved the protein-altering variants (1.2% vs 0.2%, SKAT-O, P < 0.0001). Interestingly, 19 variants were predicted to be damaging or had been previously reported in PD or DRD patients (defined as “deleterious”, Fig. 2), of which significant associations were detected (1.2% vs 0.1%, SKAT-O, P < 0.0001) in the WES cohort (Table 1). In the cohort WGS, however, no association was observed for either all variants (1.0% vs 0.9%, SKAT-O, P = 0.70) or deleterious variants (0.5% vs 0.2%, SKAT-O, P = 0.30) (Table 1).

Schematics of GCH1 and its triphosphate cyclohydrolase 1. Synonymous variants (green), previously reported variants associated with PD or DRD (red), newly described variants with predicted damaging functional alterations (blue) and variants with predicted tolerable functional alterations (black)
Non-coding variants
Non-coding variant analysis was only conducted in the WGS cohort and a total of 1702 variants (including those in the regulatory elements, eQTLs, UTRs/introns and GWAS signals, respective numbers of which are shown in Table S1) were identified, including 1486 rare variants and 216 common variants.
Among the 216 common variants (Table S3), we identified that rs12323905 remained significantly associated with PD after multiple comparison correction in our data (odds ratio = 1.19, 95%CI 1.07–1.32, P = 0.001). The previously reported associated variants rs841 (odds ratio = 0.87, 95%CI 0.78–0.97, P = 0.01) and GWAS signal rs11158026 (odds ratio = 1.17, 95%CI 1.06–1.3, P = 0.003) were also found to be associated with PD in the same association direction [9, 12], however, these associations did not remain significant after multiple comparison correction. Of note, both rs11158026 (r2 = 0.98) and rs841 (r2 = 0.34) were linked to the top significant variant rs12323905 in our dataset (Fig. 3).

Target GCH1 regions with associative results and linkage disequilibrium. The top scatter plot shows the association analysis of P-values in the WGS cohort and degree of linkage disequilibrium (LD) with the lead single variant. The solid line marks the Bonferroni-corrected significance threshold and the dashed line marks the suggestive significance threshold of P < 0.05. In the target regions, gray color depicts the range of eQTLs, blue color depicts regulatory regions, and orange shows transcript region. The bottom plot demonstrates the LD blocks of target regions. *Three variants were not included in the figure because these were in the distal region of present targets and were not associated with PD.
To determine which sets of GCH1 non-coding variants were associated with the PD risk, we performed exploratory analyses using SKAT-O and SKAT-C to assess the potential contribution of variants within different sets considered independently. In the collapse analysis of rare variants, variants from both UTR/intron regions (SKAT-O, P = 0.80) and eQTLs (SKAT-O, P = 0.18) did not show any significant association with PD, whereas one of the promoter enhancer sets demonstrated suggestive difference between PD patients and controls (GH14J054857, SKAT-O, P = 0.03). Moreover, suggestive differences were observed in combinations of common and rare variants from each variant set. The UTR/intron regions (SKAT-C, P = 0.03), eQTLs (SKAT-C, P = 0.02) and a regulatory region (GH14J054880, SKAT-C, P = 0.02) were detected with significant differences (Table 2). However, none of them remained significant after multiple comparison correction.
Genotype-phenotype correlation
Next, we analyzed the phenotypic characteristics of GCH1 variant-carriers. We found that patients with deleterious variants had younger AAO than non-carriers in the WES cohort (P < 0.0001, Table 3), and the suggestive association was even observed in the WGS cohort (P = 0.048, Table 4) where the burden of deleterious variants did not significantly differ from the controls. No significant association was found between rs12323905 and AAO in the WGS cohort. In addition, patients with deleterious variants in the WES cohort showed milder motor symptoms (UPDRS II, P = 0.022; UPDRS III, P = 0.025), reduced bradykinesia symptoms (P = 0.024), and less severe fatigue scale (P = 0.035) with suggestive significance (Table 3), which suggested that deleterious variants were not only related to the risk of PD, but also suggestively associated with the alleviated motor symptoms. Likewise, in the WGS cohort, we observed suggestive association between deleterious variants and more autonomic nervous dysfunction, as well as between rs12323905 and lesser fatigue symptoms.
Meta-analysis of GCH1 deleterious variants and AAO
The workflow of the meta-analysis is shown in Fig. 4a. A total of 51 GCH1 deleterious variant-carriers and 6874 PD patients from six studies (including the present study) were included to analyze the association between GCH1 status and AAO in PD patients. The characteristics of the included studies [8, 9, 14, 43, 44] are shown in Fig. 4b. The funnel plot obtained was showed in Fig. 4c. The AAO in GCH1 deleterious variant-carriers was more than 6 years earlier than non-carriers [MD: − 6.42 (− 10.20, − 2.64); P = 0.0009; Fig. 4b]. Of note, meta-analysis conducted after exclusion of our study data showed borderline significance [MD: − 4.67 (− 9.53, 0.20); P = 0.06].

Meta-analysis of GCH1 deleterious variants and AAO. a Flowchart showing the search strategy and selection criteria of literature for meta-analysis; b Forest plot of AAO of PD in GCH1 deleterious variant-carriers and non-carriers; c Funnel plot of AAO of PD in GCH1 deleterious variant-carriers and non-carriers
Discussion
Over the past decades, investigations of coding and non-coding genome in PD have identified over 20 disease-causing genes and about 90 risk loci in PD [12, 22, 45]. However, most of them were identified in European ancestry populations, while little is known about the genetics of PD in other populations. With the decreased sequencing cost and development of new high-throughput technologies, systematic genetic studies in other populations have become possible. In this study, we identified that both GCH1 coding and non-coding variants are involved in PD. We found that the deleterious variants could not only increase the risk of PD, but also advance the AAO of PD by over 6 years. We also identified that rs12323905, UTRs/introns, eQTLs, and two regulatory regions may be associated with PD and the GCH1 deleterious variants may influence PD manifestations.
A total of 19 and 12 protein-altering variants of GCH1 were detected in cohorts WES and WGS, respectively. Apart from the earlier reported variants, we also found several novel missense or loss-of-function variants of GCH1 in our PD cohort, including p.I193M (n = 2), p.F122Ifs*1, p.M102L, p.Q87Sfs*29, p.P86S, p.P39L, and p.E11A (n = 1 each), of which p.I193M, p.F122Ifs*1, p.Q87Sfs*29, and p.P86S were predicted to be deleterious in our in silico analysis (Table S5). Increased burden of deleterious variants in GCH1 gene was detected in PD patients of the WES cohort, but not in the late-onset patients of the WGS cohort, which was in line with previous studies on late-onset PD [13], suggesting that the pathophysiology of PD with early-onset or familial history may differ from that of the late-onset PD. This is also supported by the observation that the incidence and prevalence of late-onset PD rapidly increased with age, whereas the incidence of early-onset PD is low [46].
To our knowledge, this is the first study to systematically analyze the association between GCH1 non-coding variants and PD risk using different sequencing techniques, especially WGS. Of note, the unreported rs12323905 was found to be significantly associated with PD in Chinese population. To date, the most significant variant in GCH1 locus revealed by GWAS in European populations was rs11158026 [11, 12]. The difference in top significant variants between different populations indicates the genetic heterogeneity among PD populations; and combination of results from different populations could provide insights into the mapping of true causal variants in GCH1 locus. Overall, our study confirmed that the GWAS signal reported in European populations has significance for Chinese populations as well, but there is a difference in the top significant variant, which warrants further functional studies to determine these associations in PD. Furthermore, our study revealed that variants in GH14J054857, GH14J054880, UTRs/introns and brain-specific eQTLs sets of GCH1 may play a role in PD pathogenesis, especially the rare variants in GH14J054857 sets. The suggestive correlations still need to be further replicated and explored. Meanwhile, the suggestive results of GCH1 non-coding variants may highlight a possible link between long-lasting alterations in dopamine synthesis and an increased risk of PD development [6]. Further mechanistic studies may provide insights into the basis of common diseases along with a potential explanation for how GCH1 correlates with the risk of PD development.
In the WES cohort, genotype-phenotype analysis showed that the carriers of deleterious variants had younger AAO, which indicates the role of GCH1 in the increased risk of PD as well as in altered AAO in PD patients. However, there was no significant difference between common variants of GCH1 and AAO, which was in line with a recent largest AAO GWAS study [47]. In addition, we did a meta-analysis of 6874 PD patients including 51 GCH1 carriers from 6 studies to evaluate the potential association between the GCH1 deleterious variants and AAO of the patients, which demonstrated that the GCH1 deleterious variants could modify the AAO in PD patients (more than 6 years ahead). Due to the limited number of PD patients carrying GCH1 deleterious variants, the association of the GCH1 deleterious variants with AAO only showed broadline significance, when the current study was not included. Furthermore, these are some preliminary suggestive findings between GCH1 variants and clinical manifestations in PD such as milder motor symptoms and more autonomic nervous dysfunction in deleterious variant-carriers. Several studies [8, 43] simply described the specific symptoms of PD patients carrying each GCH1 variant without a quantifying scale and comparisons. In our study, we used a specific rating scale to quantify the symptom severity and systematically compared the phenotypic difference between GCH1 variant-carriers and non-carriers. Given that the suggestive associations were not significant after Bonferroni correction and might be biased due to the insufficient number of GCH1 variant-carriers, these findings need further explorations in future.
There were several limitations in this study. First, the copy number variations encompassing the GCH1 locus were not under the scope of the analysis. Second, we did not explore the association of non-coding variants in subjects from cohort WES, given that the WES method could not fully detect the non-coding variants. Third, further studies to identify the functional mechanisms are warranted, including how these associated factors affect the functional roles of GCH1 and how changes in GCH1 can lead to PD pathogenesis. Fourth, studies focused on multiple aspects such as epistasis and environmental factors are also needed. Fifth, the late-onset PD patients were detected with low-coverage WGS; although we have filtered the variants through reasonable quality control, it is still worth further replication in other cohorts.
Conclusions
In conclusion, deleterious variants of GCH1 and non-coding signals especially rs12323905 were found to be associated with PD in Chinese mainland population. Clinically, rare or common variants of GCH1 may modify the phenotype of PD patients in motor and non-motor aspects, respectively. Meta-analysis further demonstrated that the rare deleterious variants of GCH1 could modify the AAO of PD. Our results highlight the importance of GCH1 locus in the risk of PD development as well as in the AAO of PD, and provide reference for experimental study design to elucidate the mechanisms of GCH1 involvement in the pathogenesis of PD.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- PD:
-
Parkinson’s disease
- DRD:
-
Dopa-responsive dystonia
- GCH1 :
-
Guanosine triphosphate cyclohydrolase 1
- GWAS:
-
Genome-wide association studies
- eQTLs:
-
Expression quantitative trait loci
- AAO:
-
Age at onset
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing
- UTRs:
-
Untranslated regions
- PD-MDCNC:
-
Parkinson’s Disease & Movement Disorders Multicenter Database and Collaborative Network in China
- MDS:
-
Movement Disorders Society
- GTEx:
-
Genotype-Tissue Expression
- MAF:
-
Minor allele frequencies
- SKAT:
-
Sequence kernel association test
- MD:
-
Mean difference
- CI:
-
Confidence intervals
- SD:
-
Standard deviation
References
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
Pang SY, Ho PW, Liu HF, Leung CT, Li L, Chang EES, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson's disease. Transl Neurodegener. 2019;8:23.
Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol. 2003;54(Suppl 6):S32–45.
Lee WW, Jeon BS. Clinical spectrum of dopa-responsive dystonia and related disorders. Curr Neurol Neurosci Rep. 2014;14:461.
Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain. 2003;126:1279–92.
Lewthwaite AJ, Lambert TD, Rolfe EB, Olgiati S, Quadri M, Simons EJ, et al. Novel GCH1 variant in Dopa-responsive dystonia and Parkinson's disease. Parkinsonism Relat Disord. 2015;21:394–7.
Thony B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat. 2006;27:870–8.
Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain. 2014;137:2480–92.
Rudakou U, Ouled Amar Bencheikh B, Ruskey JA, Krohn L, Laurent SB, Spiegelman D, et al. Common and rare GCH1 variants are associated with Parkinson's disease. Neurobiol Aging. 2019;73:231 e231–6.
Xu Q, Li K, Sun Q, Ding D, Zhao Y, Yang N, et al. Rare GCH1 heterozygous variants contributing to Parkinson's disease. Brain. 2017;140:e41.
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017;49:1511–6.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–102.
Rengmark A, Pihlstrom L, Linder J, Forsgren L, Toft M. Low frequency of GCH1 and TH mutations in Parkinson's disease. Parkinsonism Relat Disord. 2016;29:109–11.
Yan YP, Zhang B, Shen T, Si XL, Guo ZY, Tian J, et al. Study of GCH1 and TH genes in Chinese patients with Parkinson's disease. Neurobiol Aging. 2018;68:159 e153–6.
Karnuta JM, Scacheri PC. Enhancers: bridging the gap between gene control and human disease. Hum Mol Genet. 2018;27:R219–27.
Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging. 2016;37:209 e201–7.
Liang D, Shu L, Pan H, Xu Q, Guo J, Yan X, et al. Clinical characteristics of PD patients with LRRK2 G2385R and R1628P variants. Neurosci Lett. 2018;685:185–9.
Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson's disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord. 2018;33:730–41.
Zhang Y, Shu L, Zhou X, Pan H, Xu Q, Guo J, et al. A meta-analysis of GBA-related clinical symptoms in Parkinson's disease. Parkinsons Dis. 2018;2018:3136415.
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30:1591–601.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. 2019;19:170–8.
Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. 2019;56:265–70.
Guo JF, Zhang L, Li K, Mei JP, Xue J, Chen J, et al. Coding mutations in NUS1 contribute to Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115:11567–72.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
Li J, Shi L, Zhang K, Zhang Y, Hu S, Zhao T, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018;46:D1039–48.
Li J, Zhao T, Zhang Y, Zhang K, Shi L, Chen Y, et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018;46:7793–804.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Zweig AS, Karolchik D, Kuhn RM, Haussler D, Kent WJ. UCSC genome browser tutorial. Genomics. 2008;92:75–84.
Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017;2017:bax028.
Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Lill CM, Mashychev A, Hartmann C, Lohmann K, Marras C, Lang AE, et al. Launching the movement disorders society genetic mutation database (MDSGene). Mov Disord. 2016;31:607–9.
Dobricic V, Tomic A, Brankovic V, Kresojevic N, Jankovic M, Westenberger A, et al. GCH1 mutations are common in Serbian patients with dystonia-parkinsonism: challenging previously reported prevalence rates of DOPA-responsive dystonia. Parkinsonism Relat Disord. 2017;45:81–4.
Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabrol B, et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain. 2009;132:1753–63.
Bandmann O, Nygaard TG, Surtees R, Marsden CD, Wood NW, Harding AE. Dopa-responsive dystonia in British patients: new mutations of the GTP-cyclohydrolase I gene and evidence for genetic heterogeneity. Hum Mol Genet. 1996;5:403–6.
Cao L, Zheng L, Tang WG, Xiao Q, Zhang T, Tang HD, et al. Four novel mutations in the GCH1 gene of Chinese patients with dopa-responsive dystonia. Mov Disord. 2010;25:755–60.
Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25:603–5.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–75.
Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841–53.
Alves G, Larsen JP, Emre M, Wentzel-Larsen T, Aarsland D. Changes in motor subtype and risk for incident dementia in Parkinson's disease. Mov Disord. 2006;21:1123–30.
Yoshino H, Nishioka K, Li Y, Oji Y, Oyama G, Hatano T, et al. GCH1 mutations in dopa-responsive dystonia and Parkinson's disease. J Neurol. 2018;265:1860–70.
Guella I, Sherman HE, Appel-Cresswell S, Rajput A, Rajput AH, Farrer MJ. Parkinsonism in GTP cyclohydrolase 1 mutation carriers. Brain. 2015;138:e349.
Pang SY, Teo KC, Hsu JS, Chang RS, Li M, Sham PC, et al. The role of gene variants in the pathogenesis of neurodegenerative disorders as revealed by next generation sequencing studies: a review. Transl Neurodegener. 2017;6:27.
Ascherio A, Schwarzschild MA. The epidemiology of Parkinson's disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–72.
Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrom L, et al. Parkinson's disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord. 2019;34:866–75.
Acknowledgements
The authors thank the patients for their participation in this study. We thank Tao Wang, PhD, MD (Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology); Ling Chen, PhD, MD (Department of Neurology, The First Affiliated Hospital, Sun Yat-sen University); Hui-fang Shang, PhD, MD (Department of Neurology, West China Hospital, Sichuan University); Wei-guo Liu, PhD, MD (Department of Neurology, Affiliated Brain Hospital of Nanjing Medical University); Piu Chan, PhD, MD (Department of Neurobiology, Xuanwu Hospital of Capital Medical University); Zheng Xue, PhD, MD (Department of Neurology, Tongji Hospital, Tongji Medical college, Huazhong University of Science and Technology); Qing Wang, PhD, MD (Department of Neurology, Zhujiang Hospital of Southern Medical University); Li Guo, PhD, MD (Department of Neurology, The First Affiliated Hospital of Jinan University); Xue-jing Wang, PhD, MD (Department of Neurology, The First Affiliated Hospital of Zhengzhou University); Chang-shui Xu, PhD, MD (Department of Neurology, Henan provincial people’s hospital); Zhen-tao Zhang, PhD, MD (Department of Neurology, Renmin Hospital of Wuhan University); Tao Chen, PhD, MD (Department of Neurology, Hainan General Hospital) from cooperating centers of PD-MDCNC for data collection. No compensation was received.
Funding
This study was supported by the National Key Research and Development Program of China (2016YFC1306000, 2017YFC0909100, 2018YFC1312000, and 2016YFC1306501) to G.J.F, T.B.S and Y.X.X, the Central Public-Interest Scientific Institution Basal Research Fund of Chinese Academy of Medical Sciences (2018-12 M-HL-025) to G.J.F, the National Natural Science Foundation of China (81873785; 81974202) to G.J.F and T.B.S, and Science and Technology Major Project of Hunan Provincial Science and Technology Department (2018SK1030) to G.J.F, the innovative team program from Department of Science & Technology of Hunan Province (2019RS1010) to G.J.F, and the Innovation-driven Team Project from Central South University (2020CX016) to G.J.F.
Author information
Authors and Affiliations
Contributions
PHX, ZYW contributed equally to the work as coauthors. GJF, TBS had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: PHX, ZYW, LJC, GJF, and TBS. Acquisition, analysis, or interpretation of data: PHX, ZYW, MJP, FZH, WYG, ZX, ZYJ, ZR, ZKL, JL, ZQ, HY, WZ, LZH, XQ, SQY, YY, HYC, CYS, DJ, LLF, ZHN, WCY, YXX, SL, JH, and TJQ. Drafting of the manuscript: PHX, ZYW, GJF, TBS. Critical revision of the manuscript for important intellectual content: All authors. Obtained funding: TBS and GJF. Administrative, technical, or material support: TJQ, LJC, TBS, and GJF. Study supervision: LJC, TBS, and GJF. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent for participation in the genetic research, which was approved by relevant oversight committees and institutional review boards.
Consent for publication
Not applicable.
Competing interests
The authors report no actual or potential conflicts of interest.
Supplementary information
Additional file 1 Table S1.
Targeted GCH1 regions and variants included in the study.
Additional file 2 Table S2.
The quality control of the targeted GCH1 regions.
Additional file 3 Table S3.
Association analysis of common non-coding variants identified in this study.
Additional file 4 Table S4.
Summary of clinical features of the Parkinson’s disease patients in this study.
Additional file 5 Table S5.
Coding variants of GCH1 identified in two cohorts.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pan, Hx., Zhao, Yw., Mei, Jp. et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson’s disease: a two-cohort case-control study. Transl Neurodegener 9, 31 (2020). https://doi.org/10.1186/s40035-020-00212-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-020-00212-3