
Abstract
The senescence-associated secretory phenotype (SASP) is a generic term for the secretion of cytokines, such as pro-inflammatory factors and proteases. It is a crucial feature of senescent cells. SASP factors induce tissue remodeling and immune cell recruitment. Previous studies have focused on the beneficial role of SASP during embryonic development, wound healing, tissue healing in general, immunoregulation properties, and cancer. However, some recent studies have identified several negative effects of SASP on fracture healing. Senolytics is a drug that selectively eliminates senescent cells. Senolytics can inhibit the function of senescent cells and SASP, which has been found to have positive effects on a variety of aging-related diseases. At the same time, recent data suggest that removing senescent cells may promote fracture healing. Here, we reviewed the latest research progress about SASP and illustrated the inflammatory response and the influence of SASP on fracture healing. This review aims to understand the role of SASP in fracture healing, aiming to provide an important clinical prevention and treatment strategy for fracture. Clinical trials of some senolytics agents are underway and are expected to clarify the effectiveness of their targeted therapy in the clinic in the future. Meanwhile, the adverse effects of this treatment method still need further study.
Similar content being viewed by others
Introduction
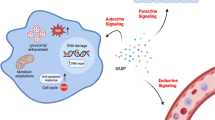
The phenomenon of cellular senescence in vitro was first reported in 1961 by Leonard Hayflflick [1], who found that human fibroblasts could not continue to expand after multiple passages, although they were still metabolically active. This contradicts the idea put forward by Carrel that cell tissue could survive permanently in vitro culture [2]. In recent decades, much progress has been made in the research field of cellular senescence, and many interesting biological phenomena can be attributed to cellular senescence. Many diseases were found associated with the accumulation of senescent cells, including cancer, atherosclerosis, and osteoarthritis. Senescence-associated secretory phenotype (SASP) is a generic term for bioactive molecules secreted by senescent cells that induce inflammation through autocrine and paracrine pathways and transmit senescence signals to neighboring cells, exacerbating telomere dysfunction and accelerating cellular senescence through tissue microenvironment, ROS-mediated pathways.
Studies have noticed the role of SASP during fracture healing. An analysis of public data displayed that markers of aging and SASP do increase during fracture healing and that fracture healing time is reduced after using drugs in mice to remove senescent cells [3]. One study reported that accelerated fracture healing was observed in aged mice after partial neutralization of TGF-β with TGF antibodies [4]. In several studies, the modulatory role was found to be displayed at systemic and cellular levels [5,6,7,8]. This literature aims to speculate on the effect of SASP produced by senescent cells on fracture healing.
Senescence and SASP
Cellular senescence and SASP
Cellular senescence is the cellular response to apoptosis, metabolic changes, and exogenous or endogenous stress. During cellular senescence, the stress or damage response can be triggered by telomere shortening, oxidative stress, oncogene activation, and DNA damage that causes cells to enter a largely irreversible cell cycle arrest and ignore apoptotic signals.
Senescent cells are still metabolically active, and most of them can secrete many cytokines, chemokines, and other bioactive molecules, collectively known as SASP or SMS [9]. Although many studies have identified SASP components in different cell types, the exact composition of SASP remains elusive and is the subject of ongoing research. However, SASP can occur differently depending on the type of senescent cells, aging triggers, and changes over time [9, 10]. SASP includes a variety of bioactive factors, which can enhance self-senescence and affect the local microenvironment of senescent cells or even the whole individual [11, 12]. SASP factors reinforce growth arrest in an autocrine manner and alter the behavior of surrounding cells in a paracrine manner [11]. Hundreds of SASP have been reported, among which the most common of factors include interleukin IL-1a, IL-6, and IL-1, and others such as IFN-γ, VEGF, ICAM-1, and GM-CSF [10, 13, 14], and some are released in the form of the exosome [14, 15].
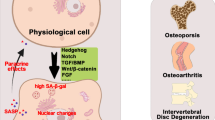
SASP is one of the typical features of senescent cells. Identification of SASP can be used as one of the methods to assess cellular senescence. The next section reveals the methodologies in detail. Although there are some qualitative and quantitative differences in SASP in different tissues and aging models, however, based on the different modes of action of SASP activity, SASP can be classified into three types, receptor-requiring (IL-1, IL-1β, IL-6, IL-8, CXCL-1, CXCL-3, CXCL-10, HGF, TGF-β, GM-CSF, etc.); direct-response (MMPs, ROS, etc.); and regulatory (TIMPs, IGFBPs, etc) [10, 12, 14, 16,17,18]. There are also some SASP released as soluble molecules or exosomes. SASP has many functions, both beneficial and harmful, as shown in Table 1. Targeting senescent cells and their SASP may provide a novel strategy for the treatment of age-related diseases, such as Alzheimer’s disease (AD). These methods of using drugs to antagonize the harmful extracellular impact of senescent cells are called senomorphics [11, 19, 20]. The specifics of this study are detailed below.
Validation of senescent cells
Senescent cells have multiple phenotypes, which vary depending on their origin, but cells can be identified based on common characteristics. Due to the phenotypic heterogeneity, senescent cells cannot be identified with only one marker, but a combination of multiple senescent cell markers is needed. Some studies suggest validating at least three different traits (Fig. 1), including the (a) arrest of cell cycle progression, (b) associated structural changes, and (c) other traits known to be specific to the senescent cells being experimented with [9]. Cells can be identified as being in proliferative arrest by detecting activation of the p16–pRB axis, p53–p21 axis, or cellular proliferation and DNA replication assays. One of the characteristics of senescent cells is an increase in lysosomal content, resulting in lysosomal β-galactosidase (β-gal) activity (also known as senescence-associated β-gal or SA-β-gal. [9]. 5-Bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) is the most common substrate for SA-β-Gal activity, which is catalyzed by SA-β-Gal to galactose and 5-bromo-4-chloro-3-hydroxyindole-1, which then dimerizes to form the blue precipitate indigo [21]. Senescent cells can also be identified by detecting DNA segments with chromatin alterations reinforcing senescence (DNA–SCARS), ROS, or telomere shortening. Cellular senescence can also be demonstrated by explicitly identifying the secretion of relevant SASP molecules. Interestingly, studies have found that macrophages can also express p16INK4a and SA-β-Gal in response to immune responses [22]. To identify senescent cells more accurately, multiple assays need to be used. If future studies can identify unique or standard markers of senescent cells, this will accelerate the development of cellular senescence-related research. Current fluorescent tracers and advanced imaging tools allow real-time monitoring of the aging process in vivo and real-time assessment of therapeutic effects [23,24,25,26]. A paper reports on the design of a two-parameter recognition fluorescent probe for precise imaging of cellular senescence that allows high-contrast imaging of senescence independent of cell source or type of stress [27]. These will enable the better translation of research results into clinical applications.

DDR occurs after DNA damage, leading to cellular senescence. Senescent cells have several features: upregulation of the BCL-2 anti-apoptotic protein family (induces resistance to apoptosis, cells undergo oxidative damage (elevated ROS can be detected), metabolic changes (including the presence of SA-β-gal aggregates, SAHF and SASP), cell cycle arrest (p21 and p16 upregulation). Osteoblasts senesce in response to stress stimuli, and these cells likely cause an inflammatory microenvironment in bone by secreting SASP effects that disrupt bone formation and enhance osteoblast function. SASP can promote aggregation of BMSCs, which can differentiate into pre-osteoblasts and osteoblasts. HSCs can differentiate into osteoclasts BMSCs, bone-marrow-derived mesenchymal stem cells. DDR, DNA damage response. RANK, receptor activator of nuclear factor Kappa-B. RANKL, RANK ligand. ROS, reactive oxygen species. SAHF, senescence-associated heterochromatin foci. SASP senescence-associated secretory phenotype, SA-β-gal senescence-associated β-galactosidase. HSC haematopoietic stem cell
The regulation of SASP
The regulation of SASP is complex and involves many factors. IL-1a is an upstream regulator of other SASP and regulates the secretion of other SASP [8, 16, 28]. NF-κB signalling regulates the senescence-associated secretory phenotype (SASP) and together with the transcription factor C/EBPβ, co-activates promoters of SASP genes. Some SASP can enhance the transcription of SASP such as IL-1a, IL-6, and IL-8 by regulating the activities of NF-κB and CEBPβ. IGFBP3, IGFBP4, and IGFBP7 are critical players in SASP and are thought to mediate cellular senescence through paracrine signaling [5,6,7,8].
The DNA damage response and SASP
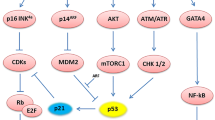
The accumulation of DNA damage caused by replicated cells can cause DNA damage response (DDR), which can directly cause cell cycle arrest. DDR can also induce cell senescence and secrete SASP by activating two major kinase systems, ataxia telangiectasia mutant (ATM) and ATM and rad3-related (ATR), which ultimately phosphorylate p53 [29]. ATM and/or ATR are well-known upstream molecules of checkpoint kinase (CHK)1/CHK2. Stress-induced activation of MAPK and p38 was also shown to be sufficient to trigger cell senescence and SASP [30].
Regulation pathway of SASP translation
Nuclear factor κB (NF-κB) is an essential component in the regulatory pathway of SASP. The regulatory effects of ATM, IL-1a, and p38 on SASP are achieved by changing the activity of NF-κ B [11, 31]. NF-κB can bind to the transcription factor CCAAT/enhancer-binding protein-β (CEBPβ) and activate the promoter of the SASP gene [32], such as those that encode chemokines. The transcription factor GATA 4 can activate some genes of SASP, including IL-6, IL-8, CXCL 1(GROα), C-CSF, and ECM protease [33]. As an intermediate mediator of DDR signals and NF-κB activation, GATA 4 can regulate NF-κB activity by increasing the expression and secretion of IL-1 A and ultimately affect the secretion of SASP [34]. JAK/STAT pathway is highly activated in senescent cells and is one of the main ways to regulate cytokine production, which is related to the expression of IL-6, IL-8, IL-1A, CXCL-1, CXCL-2, CXCL-5, CXCL-6 and CXCL-10 [35]. HMGB factors constitute a family of non-histone architectural proteins that contain a distinct DNA-binding domain. HMGB2 directly binds and precisely regulates the expression of the SASP gene in oncogene-induced senescent cells [36]. The mTOR mediates cell responses to stresses, such as DNA damage and nutritional deficiencies. mTOR can activate the NF-κB pathway by promoting the translation of IL-1 A, thus affecting the secretion of SASP, including IL-6 and IL-8. On the other hand, the transcription of many SASP factors is influenced by the MAPKp38 pathway [37].
The innate immunity and SASP
cGAs (cGMP–AMP synthase) is a DNA sensor located in the cytoplasm of non-splinter cells, which can activate the innate immune response and lead to cell senescence. The absence of cGAs accelerated the spontaneous immortalization of mouse embryonic fibroblasts and eliminated SASP induced by natural aging and DNA damage agents [38, 39]. The activated cGAs generate a second messenger ring nucleotide (cGAMP) that binds to the stimulator of interferon genes (STING) and promotes the aggregation of Sting with TANK-binding kinase 1(TBK 1) and IκB kinases. Therefore, interferon regulatory factor 3(IRF3) and NF-κB are activated, respectively, resulting in the production of type I interferon and SASP factors (such as IL-6, IL-8, IL-1 β, and MMP-12) [7, 40, 41].
Other regulation methods
Recent studies have documented that extracellular nicotinamide phosphoribosyltransferase (ENAMPT) is also a component of SASP and is related to the activation of p53/p21 by NAD/NADH caused by mitochondrial damage, leading to cell senescence [42]. It has been confirmed that high-mobility group box 1protein (HMGA1) is involved in the regulation of the secretion of pro-inflammatory SASPs (IL-6, IL-8, etc.) through the regulation of NAMPT and ultimately the NAD/NADH ratio [43], and that pro-inflammatory SASP has a pro-tumorigenic effect [44]. Moreover, in mouse embryos overexpressing the HMGA1 P6 pseudogene, the mice grew faster and aged later [45]. These are associated with the activation of the p38MAPK pathway [43, 46]. The secretion of SASP can be regulated using components of the drug action regulation pathway to treat diseases, such as tumors, diabetes, and tendinopathy [11, 47].
The inflammation of fracture healing in generally population
Bone is composed of bone extracellular matrix proteins, inorganic minerals, and a variety of resident cell types. It is an essential part of the locomotor system [48]. There are two methods of fracture healing: direct (primary) and indirect (secondary). Direct fracture healing is rare, and the fracture end needs to meet the conditions of anatomic reduction and complete the structural remodeling through direct intramembranous osteogenesis without forming calluses. Indirect fracture healing is the most common form, including endochondral and intramembranous ossification. According to the time sequence, fracture healing can be divided into hematoma formation, fibrocartilaginous callus formation, bony callus formation, and bone remodeling [49].
Inflammation at the fracture healing site
During the early course of fracture healing, hematomas form, and in the first 48 h, an inflammatory response occurs at the fracture site, manifested by invasion of macrophages, polymorphonuclear leukocytes, and lymphocytes. After the injury, a large amount of danger-associated molecular pattern (DAMP) appears. The ruptured vasculature and exposed bone marrow cause inflammatory cell infiltration at the injured site, and fracture-related hematoma (dense cell mass) is formed by a variety of inflammatory cells (neutrophils, macrophages, T cells, B cells, regulatory B-cell mast cells.) and platelets and red blood cells [50]. The hematoma helps to initiate healing and provides the foundation for bone tissue formation. The loss of hematoma may delay the healing of the fracture. Mesenchymal stromal cells (MSCs) and hematopoietic stem cells have multidirectional differentiation potential and are closely associated with fracture healing.
Inflammatory cells, such as granulocytes and macrophages, secrete a variety of cytokines and growth factors (IL-1, IL-6, TNF-α, inducible nitric oxide synthase, transforming growth factor-β, platelet-derived growth factor, insulin-like growth factor, and fibroblast growth factor 2) [51], which are an essential part of the signaling environment for fracture healing. First, under the action of proinflammatory mediators (such as growth factors, cytokines, and chemokines), neutrophils gather at the injury site after fracture and further secrete cytokines to aggregate monocytes [52]. At the same time, activated macrophages appear, peaking at 3–7 days, releasing cytokines such as IL-1β and TNF-α to stimulate fibroblast proliferation and attract other marrow cells and lymphocytes to gather at the injury site [53]. Macrophages eventually differentiate into osteoclasts under RANKL and M-CSF and are involved in fracture repair [54]. The RANKL is necessary and sufficient for the induction of osteoclast differentiation and function, and MCSF induces osteoclast proliferation. Increased osteoclast formation accelerates cartilage resorption and promotes osseointegration. Inflammatory macrophages are essential to initiate and propagate endochondral osteogenesis [55]. Macrophages are the primary immune cells that initiate and maintain the inflammatory response and are directly involved in the osteogenesis process by secreting important cytokines associated with osteogenesis. Besides participating in allergic reactions and autoimmunity, mast cells also promote wound and fracture healing [56]. They can stimulate blood vessel permeability and angiogenesis and regulate bone metabolism [57]. The histamine and VEGF induce hyperpermeability of injured blood vessels and provide a suitable environment for tissue repair [58]. Ragipoglu et al. speculated that the effect of mast cells on bone healing might be related to their recruitment of vascular endothelial cells and coordination of metabolic activities [59].
Osteoblasts and osteoclasts have been reported to have direct cell-to-cell contact with lymphocytes, indicating the regulatory role of immune cells in the late stage of fracture healing [60]. T cells can secrete RANKL to activate osteoclasts. Interestingly, the relative number of CD4( +) T cells and CD8( +) T cells changes after fracture [60]. This could be a mechanism to enhance the bone healing process in the injured bone as CD4( +) T cells have been reported to have a pronounced osteogenic role [61, 62]. Another T-cell subset, Th17 cells, secretes IL 17F to promote osteoblast growth. On the other hand, another T-cell subset (Tregs) can inhibit the function of osteoblasts and osteoclasts by secreting IL-4, IL-10, and TGF-β [63]. γδT cells, also known as inflammatory lymphocytes, respond to acute inflammatory stress signals, promote cytokine production, recruit macrophages[64], and promote the formation of osteoblasts. It has been found that the secretion of IL17A by γδT cells can stimulate the proliferation of mesenchymal progenitor cells and the differentiation of osteoblasts [65].
B cells secrete osteoprotegerin (OPG). The protein is also expressed in osteoblasts to regulate the activity of RANKL [66]. B cells positively affected osteoclasts, whereas CD8 T cells had the opposite effect. By observing the expression ratio of OPG and RANKL, Choi et al. judged the infiltration degree of T cells and B cells during bone healing and then inferred the specific stage of the healing process [67]. Regulatory B cells (Bregs) are a kind of B cells that promote endogenous bone regeneration in the initial healing stage [68]. Bregs are a newly discovered B-cell subset that promotes endogenous bone regeneration in the initial healing phase [69]. Bregs can secrete IL-10. Studies have proved that delayed healing patients downregulated B-cell IL-10 secretion early and Bregs dysfunction may be one of the reasons for delayed fracture healing [69].
Fracture healing is achieved through the interaction and crosstalk of stem cells, immune cells, and bone cells
Direct fracture healing refers to the direct differentiation of MSCs into functional anabolic bone cells [70]. In indirect fracture healing, MSCs are transformed into chondroblasts regulated by transcription factor SRY-related high mobility group-box gene 9 (Sox9) and M2-type macrophages [71]. These cells will further differentiate into chondrocytes and finally transform into hypertrophic chondrocytes during the formation of hard callus [72]. These cells differentiate into osteoblasts under the induction of transcription factors, such as Runx2 and Sp7, etc. [73]. Bone-marrow mesenchymal stromal cells differentiate into osteoblast phenotype in the periosteum and surrounding soft tissues, and the Wnt/β-catenin pathway is involved in this process [74]. The osteoblast transcription factor RUNX2, the enzyme alkaline phosphatase, and other mineralization proteins can mineralize bone matrix [75]. Osteoclasts are large, multinucleate cells that secret acid and proteolytic enzymes to dissolve the bone matrix. Nuclear factor Kappa-B (RANK) ligand (RANK-L) is an anchored cell membrane factor that can interact with RANK as its receptor and eventually induce osteoclast precursor maturation [76]. Osteoblasts themselves secrete GM-CSF, which is involved in promoting osteoclast differentiation and maturation [77]. Future research will focus on understanding how multiple cell types and the resulting signaling networks integrate spatially over time to regulate healing. The inflammatory response during fracture healing is of remarkable complexity, but in silico models help us to understand the principles that regulate the various events that occur at the tissue, cellular, and subcellular levels. Silico models of the inflammatory response in bone fracture healing have been constructed to further explore the crosstalk of multiple cells in fracture healing [78].
SASP in fracture healing
Existing studies confirmed that SASP contributes to the recovery of injured tissues [79, 80]. What is the role of SASP in skeletal injury and repair? One study analyzed SASP-related components in fracture healing tissues using qRT-PCR and found that most SASP increased rapidly during fracture healing, especially CCL7 increased 70-fold at day 14 of the fracture and Plasminogen activator inhibitor-1 (Pai1 or Serpine1) increased more than 60-fold at day 8 but decreased substantially at day 14. At the same time, interleukins showed a significant increase at the beginning of fracture healing, such as IL-6 and IL-17 [3]. The variety of SASPs the high heterogeneity of gene expression of SASPs, and the timing of their peak concentrations may be related to their effects.
IL-6
Bone-marrow-derived mesenchymal stromal cells (BMSCs) play an essential role in fracture healing, differentiating into osteoblasts and BMSCs recruit to bone resorption sites for bone tissue remodeling. Animal studies found that SASP components such as IL-6 were secreted by SA-β-gal-positive, cell cycle-arrested senescent cells in irradiated mice and found that SASP further affected the differentiation impairment of BMSCs through paracrine signaling [81]. It has been suggested that IL-6 deficiency enhances the expression of osteoblast-related genes (Runx2 and Col1a1) and decreases the expression of osteoclast-related genes [82]. A study indicated that SASP targeting might be an effective treatment for irradiation-induced bone loss [81]. One study found more IL-6 from BMSCs from older adults than younger adults, and BMSCs can regulate osteoblast and osteoclast activity through the secretion of SASP [83]. IL-6 acts as an osteoblast inhibitor and promotes osteoclast formation [84], affecting bone remodeling and possibly fracture healing.
TNF-α
It was reported that TNF-α, such as IL-6, has a stimulating effect on bone resorption but an inhibitory effect on bone formation [84, 85], which may be related to the regulation of RANKL [86]. Some studies confirmed that RANKL-dependent pathways activated by pro-inflammatory cytokines can induce osteoclast formation, therefore, enhancing osteoclast activity [87, 88]. TNF-α can also indirectly increase RANKL expression and affect osteoclast differentiation by activating osteoblasts, B cells, and T cells [88]. One study in mice has found that p12 mediates the bone inhibition of TNF-α and TNF-β, and it is found that the use of TNF-α and TNF-β blockers in the aged mouse model can accelerate the healing of mouse fractures [89]. It has also been suggested that aging leads to increased long-term expression of TNF-α, which leads to delayed early inflammation and cartilage formation processes in fracture healing and a decrease in overall VEGF, which affects angiogenesis to the point of affecting bone healing, as well as being one of the main reasons why diabetes affects bone healing in mice [90]. In contrast, Glass et al. found that low concentrations of TNF-α could promote bone healing by enhancing the recruitment and differentiation of muscle-derived stromal cells [91].
TGF-β
TGF-β is also an integral component of SASP, and elevated markers of cellular senescence and enhanced TGF expression were observed at fracture sites in mice. TGF-β1 can potentially promote the recruitment of MSCs [92], which are involved in fracture healing upon differentiation. Scholars have reported that TGF-β contributes to treating bone defects in rats [93]. Some scholars have observed that bone samples from aged mice and humans have high levels of TGF-β and can stimulate the breakdown of TNF receptor-associated factor 3 to inhibit the differentiation of mesenchymal progenitor cells into osteoblasts [94]. High levels of TGF-β were detected in the blood of both mice and humans with fracture nonunion [95]. One study reported that accelerated fracture healing was observed in aged mice after partial neutralization of TGF-β with TGF antibodies [4]. However, TGF-β has also been identified to contribute to the angiogenesis of fracture-healing tissue. During osteoclastogenesis, RANKL drives osteoclast differentiation, while OPG antagonizes RANKL action, and the RANKL/OPG ratio affects the osteoclast differentiation rate and bone resorption process [96, 97]. Different concentrations of TGF-β have different effects on the RANKL/OPG ratio and ultimately on osteoclast differentiation (low concentrations of TGF-β increase the stimulation of osteoclast differentiation, while high concentrations of TGF-β have the opposite effect) [98, 99].
Other SASP factors
The differentiation process of BMSCs is mediated by IL-8, and MMP [100, 101], which are also the SASP molecules, which may affect the differentiation outcome of BMSCs, which may be one way in which SASP affects fracture healing and bone loss. The function of SASP in skeletal tissues needs further experiments. IL-8 and MMP9 may affect the activity of these cells, which are critical participating cells in the bone healing process, in the manner described above and may, therefore, influence the bone healing process (Fig. 1). There are relatively few experimental studies addressing SASP on fracture healing, and a recent study performed computer analysis of public mRNAseq data found that senescence and SASP were associated with fracture healing [3]. The currently available results demonstrate that the effects of SASP on the skeletal system are primarily detrimental. However, transient SASP was found to promote tissue recovery in wound healing and liver fibrosis. In contrast, chronic SASP had unfavorable effects on tissues [102, 103], and it has been speculated that there may be a threshold beyond which SASP would have different effects [3]. The local effects of senescence, including those of SASP, may be related to the abundance and duration of senescent cell burden. The accumulation of senescent cells leads to adverse effects, negatively correlated with local immune clearance.
However, the onset of cellular senescence in healing bone is also a short-time course, but the bone healing-promoting effect of SASP was not observed in the available experiments. This may be because the highly inflammatory state [104] after fracture masks the positive impact of transiently appearing senescent cells. On the other hand, bone, unlike other tissues, such as skin viscera, is the only tissue in the organism that is fully recoverable and does not form scars. We can then wonder whether the mechanism of SASP repair of injured tissues interacts with the mechanism of scar formation. This question needs to be addressed by further research explorations.
Senolytics and fracture healing
Numerous findings have shown that senescent cell removal is largely beneficial, leading to an exponential advance in research on therapeutic strategies for senescence depletion (called senotherapy) [105]. A variety of drugs with the ability to eliminate senescent cells have now been reported. These drugs can selectively target senescent cells (called senolytics) or selectively inhibit SASP (called senomorphics) from reducing or suppressing senescent cells in the organism [11] (Fig. 1). Some researchers have classified senolytics into three major classes: Class I senolytics are BCL-2 family protein inhibitors whose inhibition culminates in the selective apoptosis of senescent cells [106], such as ABT-737 and ABT-263 (also known as navitoclax) [11]; Class II senolytics can inhibit pro-survival signals upstream of senescent cells to resist cell death. Such as the senolytic peptide FOXO4–DRI acts by interfering with the binding of FOXO4 and p53 [107], as dasatinib and quercetin act by downregulating AKT signaling [108], HSP90 inhibitors also act as senolytics by mediating the downregulation of AKT signaling [109], and the mTOR inhibitor rapamycin acts by affecting NF-κB [110]; Class III senolytics can interfere with the intracellular homeostasis of senescent cells, such as piperlongumine, and procyanidin C(add source). Senomorphics can inhibit the extracellular function of senescent cells by targeting senescence-associated signaling pathways (e.g., MAPK, NF-κB, mTOR) while maintaining the survival of senescent cell (add source)s.
Some projects that used senolytics (dasatinib and quercetin) in aged mice with established bone loss have observed a reduction in senescent osteoclasts [111], contributing to the treatment of osteoporosis. However, some studies reported trabecular bone loss and impaired bone formation in BMSC in aged mice after using Navitoclax (ABT-263) [112]. A study reported that intermittent treatment of young fracture mice with dasatinib and quercetin resulted in a downregulation of aging markers in fracture healing tissue and contributed to fracture healing [3]. In contrast, short-term treatment of fracture mice with dasatinib and quercetin (1, 3, 5, and 7 days after fracture) revealed that accelerated fracture healing was observed only in the older mice [4]. Several clinical trials are currently underway: dasatinib and quercetin are testing for bone health (NCT04313634), fisetin is testing for skeletal health (NCT04313634), and osteoarthritis (NCT04210986).
The relationship between senolytics and fracture healing continues to be studied, and there are no precise rules for the dose and duration of treatment. Senolytics can also cause some adverse effects. Some studies have found that some senolytics may cause severe reductions in platelets or neutrophils [112, 113]. The drug causes massive apoptosis of senescent cells, which may lead to tissue atrophy [114]. Although the mechanism by which the adverse effects of senolytics occur is not yet clear, existing studies suggest that long-term use of senolytics drugs may lead to additional side effects. The safety of senolytics will be the focus of the next studies. The safety of senolytics will be the focus of further research.
Conclusions
Senescence is a state of cellular proliferative arrest as senescent cells secrete SASP and exert paracrine effects, affecting neighboring cells. The effects of senescence on fracture healing involve the four stages of fracture healing. Fracture healing is a complex process involving multiple cells and molecules, and the inflammatory response affects the quality of fracture healing. SASP can affect inflammatory molecules in fracture healing and multiple cellular components involved in healing, but the mechanisms are not yet precise. In addition, senolytics are potentially effective in treating non-healing fractures, but the specific dosing method still needs to be simpler. In conclusion, further studies are needed to investigate the effects of SASP on fracture healing and to assess whether treatment targeting SASP will promote fracture healing.
Availability of data and materials
The authors can be contacted for data requests.
Abbreviations
- ATM:
-
Ataxia telangiectasia mutant
- ATR:
-
ATM and rad3-related
- CEBP:
-
CCAAT/enhancer-binding protein
- CHK:
-
Checkpoint kinase
- CXCL:
-
C–X–C motif chemokine ligand
- DDR:
-
DNA damage response
- ECM:
-
Extracellular matrix
- EVs:
-
Extracellular vesicles
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HMGB:
-
High mobility group box
- ICAM:
-
Intercellular adhesion molecule
- IFN:
-
Interferon
- IGFBP:
-
Insulin-like growth factor binding protein
- IL:
-
Interleukin
- JAK/STAT:
-
Janus kinase/signal transducer and activator of the tran-ions
- MAPK:
-
Mitogen-activated protein kinase
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mechanistic target of rapamycin
- NF-κB:
-
Nuclear factor κB
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory phenotype
- SMS:
-
Senescence-messaging secretome
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
References
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Carrel A. On the permanent life of tissues outside of the organism. J Exp Med. 1912;15(5):516–28.
Saul D, Monroe DG, Rowsey JL, Kosinsky RL, Vos SJ, Doolittle ML, et al. Modulation of fracture healing by the transient accumulation of senescent cells. elife. 2021. https://doi.org/10.7554/eLife.69958.
Liu J, Zhang J, Lin X, Boyce BF, Zhang H, Xing L. Age-associated callus senescent cells produce TGF-β1 that inhibits fracture healing in aged mice. J Clin Invest. 2022. https://doi.org/10.1172/JCI148073.
Zhang B, Fu D, Xu Q, Cong X, Wu C, Zhong X, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. 2018;9(1):1723.
Ozcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8(7):1316–29.
Favaro F, Luciano-Mateo F, Moreno-Caceres J, Hernandez-Madrigal M, Both D, Montironi C, et al. TRAIL receptors promote constitutive and inducible IL-8 secretion in non-small cell lung carcinoma. Cell Death Dis. 2022;13(12):1046.
Alessio N, Squillaro T, Di Bernardo G, Galano G, De Rosa R, Melone MAB, et al. Increase of circulating IGFBP-4 following genotoxic stress and its implication for senescence. elife. 2020. https://doi.org/10.7554/eLife.54523.
Gonzalez-Gualda E, Baker AG, Fruk L, Munoz-Espin D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288(1):56–80.
Homann L, Rentschler M, Brenner E, Bohm K, Rocken M, Wieder T. IFN-gamma and TNF induce senescence and a distinct senescence-associated secretory phenotype in melanoma. Cells. 2022. https://doi.org/10.3390/cells11091514.
Di Micco R, Krizhanovsky V, Baker D. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22(2):75–95.
Hao X, Wang C, Zhang R. Chromatin basis of the senescence-associated secretory phenotype. Trends Cell Biol. 2022;32(6):513–26.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Chen L, Mei G, Jiang C, Cheng X, Li D, Zhao Y, et al. Carbon monoxide alleviates senescence in diabetic nephropathy by improving autophagy. Cell Prolif. 2021;54(6): e13052.
Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1): e3000599.
Byun HO, Lee YK, Kim JM, Yoon G. From cell senescence to age-related diseases: differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015;48(10):549–58.
Park SY, Kim YS, Yang DJ, Yoo MA. Transcriptional regulation of the Drosophila catalase gene by the DRE/DREF system. Nucleic Acids Res. 2004;32(4):1318–24.
Alic N, Hoddinott MP, Vinti G, Partridge L. Lifespan extension by increased expression of the Drosophila homologue of the IGFBP7 tumour suppressor. Aging Cell. 2011;10(1):137–47.
Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD, et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol. 2021;61:779–803.
Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. 2022. https://doi.org/10.1172/JCI158450.
Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–22.
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging. 2017;9(8):1867–84.
Paez-Ribes M, Gonzalez-Gualda E, Doherty GJ, Munoz-Espin D. Targeting senescent cells in translational medicine. EMBO Mol Med. 2019;11(12): e10234.
Zhen Z, Zhu S, Jin J, Wang L, Lu W. A water-soluble probe with p-hydroxybenzyl quaternary ammonium linker for selective imaging in senescent cells. Anal Chim Acta. 2020;1133:99–108.
Sinha S, Sinha A, Dongre P, Kamat K, Inamdar MS. Organelle dysfunction upon asrij depletion causes aging-like changes in mouse hematopoietic stem cells. Aging Cell. 2022;21(4): e13570.
Wagner KD, Wagner N. The senescence markers p16INK4A, p14ARF/p19ARF, and p21 in organ development and homeostasis. Cells. 2022. https://doi.org/10.3390/cells11121966.
Li J, Wang L, Luo X, Xia Y, Xie Y, Liu Y, et al. Dual-parameter recognition-directed design of the activatable fluorescence probe for precise imaging of cellular senescence. Anal Chem. 2023;95(8):3996–4004.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–90.
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9.
Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–48.
Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, et al. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-kappaB activation. Aging Cell. 2019;18(1): e12858.
Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979–92.
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612.
Mazzucco AE, Smogorzewska A, Kang C, Luo J, Schlabach MR, Xu Q, et al. Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes Dev. 2017;31(19):1933–8.
Kandhaya-Pillai R, Miro-Mur F, Alijotas-Reig J, Tchkonia T, Kirkland JL, Schwartz S. TNFalpha-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging. 2017;9(11):2411–35.
Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215(3):325–34.
Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. Author correction: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2021;23(5):564–5.
Blasiak J. Senescence in the pathogenesis of age-related macular degeneration. Cell Mol Life Sci. 2020;77(5):789–805.
Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–70.
Cao X, Li M. A new pathway for senescence regulation. Genomics Proteom Bioinform. 2015;13(6):333–5.
Nacarino-Palma A, Rico-Leo EM, Campisi J, Ramanathan A, Gonzalez-Rico FJ, Rejano-Gordillo CM, et al. Aryl hydrocarbon receptor blocks aging-induced senescence in the liver and fibroblast cells. Aging. 2022;14(10):4281–304.
Kuehnemann C, Hu KQ, Butera K, Patel SK, Bons J, Schilling B, et al. Extracellular nicotinamide phosphoribosyltransferase is a component of the senescence-associated secretory phenotype. Front Endocrinol. 2022;13: 935106.
Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 2019;21(3):397–407.
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101.
De Martino M, Palma G, Arra C, Chieffi P, Fusco A, Esposito F. Characterization of HMGA1P6 transgenic mouse embryonic fibroblasts. Cell Cycle. 2020;19(18):2281–5.
Hao W, Shan W, Wan F, Luo J, Niu Y, Zhou J, et al. Canagliflozin delays aging of HUVECs Induced by Palmitic Acid via the ROS/p38/JNK pathway. Antioxidants. 2023. https://doi.org/10.3390/antiox12040838.
Wang C, Zhou Z, Song W, Cai Z, Ding Z, Chen D, et al. Inhibition of IKKbeta/NF-kappaB signaling facilitates tendinopathy healing by rejuvenating inflamm-aging induced tendon-derived stem/progenitor cell senescence. Mol Ther Nucleic Acids. 2022;27:562–76.
Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. 2008;3(Suppl 3):S131-139.
Sheen JR, Garla VV: Fracture Healing Overview. In: StatPearls. edn. Treasure Island (FL); 2022.
Grundnes O, Reikeras O. The importance of the hematoma for fracture healing in rats. Acta Orthop Scand. 1993;64(3):340–2.
Sun G, Wang Z, Ti Y, Wang Y, Wang J, Zhao J, et al. STAT3 promotes bone fracture healing by enhancing the FOXP3 expression and the suppressive function of regulatory T cells. APMIS. 2017;125(8):752–60.
Mangum LH, Avila JJ, Hurtgen BJ, Lofgren AL, Wenke JC. Burn and thoracic trauma alters fracture healing, systemic inflammation, and leukocyte kinetics in a rat model of polytrauma. J Orthop Surg Res. 2019;14(1):58.
Leibovich SJ, Ross R. The role of the macrophage in wound repair a study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78(1):71–100.
Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, et al. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104(13):4029–37.
Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol. 2012;8(3):133–43.
Lindholm R, Lindholm S, Liukko P, Paasimaki J, Isokaanta S, Rossi R, et al. The mast cell as a component of callus in healing fractures. J Bone Joint Surg Br. 1969;51(1):148–55.
Grutzkau A, Kruger-Krasagakes S, Baumeister H, Schwarz C, Kogel H, Welker P, et al. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206. Mol Biol Cell. 1998;9(4):875–84.
Kroner J, Kovtun A, Kemmler J, Messmann JJ, Strauss G, Seitz S, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res. 2017;32(12):2431–44.
Ragipoglu D, Dudeck A, Haffner-Luntzer M, Voss M, Kroner J, Ignatius A, et al. The role of mast cells in bone metabolism and bone disorders. Front Immunol. 2020;11:163.
Konnecke I, Serra A, El Khassawna T, Schlundt C, Schell H, Hauser A, et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone. 2014;64:155–65.
Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat Med. 2011;17(12):1594–601.
Grassi F, Cattini L, Gambari L, Manferdini C, Piacentini A, Gabusi E, et al. T cell subsets differently regulate osteogenic differentiation of human mesenchymal stromal cells in vitro. J Tissue Eng Regen Med. 2016;10(4):305–14.
Nam D, Mau E, Wang Y, Wright D, Silkstone D, Whetstone H, et al. T-lymphocytes enable osteoblast maturation via IL-17F during the early phase of fracture repair. PLoS ONE. 2012;7(6): e40044.
Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100.
Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing gammadelta T cells enhance bone regeneration. Nat Commun. 2016;7:10928.
Manabe N, Kawaguchi H, Chikuda H, Miyaura C, Inada M, Nagai R, et al. Connection between B lymphocyte and osteoclast differentiation pathways. J Immunol. 2001;167(5):2625–31.
Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ, Kim HM, et al. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol. 2001;31(7):2179–88.
Shao S, Scholtz LU, Gendreizig S, Martinez-Ruiz L, Florido J, Escames G, et al. Primary head and neck cancer cell cultures are susceptible to proliferation of Epstein-Barr virus infected lymphocytes. BMC Cancer. 2023;23(1):47.
Yang S, Ding W, Feng D, Gong H, Zhu D, Chen B, et al. Loss of B cell regulatory function is associated with delayed healing in patients with tibia fracture. APMIS. 2015;123(11):975–85.
Ehnert S, Relja B, Schmidt-Bleek K, Fischer V, Ignatius A, Linnemann C, et al. Effects of immune cells on mesenchymal stem cells during fracture healing. World J Stem Cells. 2021;13(11):1667–95.
Duchamp de Lageneste O, Julien A, Abou-Khalil R, Frangi G, Carvalho C, Cagnard N, et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun. 2018;9(1):773.
Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144(2):221–34.
Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, et al. Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J Cell Biol. 2004;166(1):85–95.
Shiu HT, Leung PC, Ko CH. The roles of cellular and molecular components of a hematoma at early stage of bone healing. J Tissue Eng Regen Med. 2018;12(4):e1911–25.
Mansour A, Mezour MA, Badran Z, Tamimi F. (*) Extracellular matrices for bone regeneration: a literature review. Tissue Eng Part A. 2017;23(23–24):1436–51.
AlQranei MS, Senbanjo LT, Aljohani H, Hamza T, Chellaiah MA. Lipopolysaccharide- TLR-4 Axis regulates osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol. 2021;22(1):23.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–76.
Lafuente-Gracia L, Borgiani E, Nasello G, Geris L. Towards in silico models of the inflammatory response in bone fracture healing. Front Bioeng Biotechnol. 2021;9: 703725.
Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, Cazin C, et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. 2017;20(3):407–14.
Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–83.
Bai J, Wang Y, Wang J, Zhai J, He F, Zhu G. Irradiation-induced senescence of bone marrow mesenchymal stem cells aggravates osteogenic differentiation dysfunction via paracrine signaling. Am J Physiol Cell Physiol. 2020;318(5):C1005-c1017.
Zhu S, He H, Gao C, Luo G, Xie Y, Wang H, et al. Ovariectomy-induced bone loss in TNFalpha and IL6 gene knockout mice is regulated by different mechanisms. J Mol Endocrinol. 2018;60(3):185–98.
Siegel G, Kluba T, Hermanutz-Klein U, Bieback K, Northoff H, Schafer R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013;11:146.
Huang P, Zhang C, Delawary M, Korchak JA, Suda K, Zubair AC. Development and evaluation of IL-6 overexpressing mesenchymal stem cells (MSCs). J Tissue Eng Regen Med. 2022;16(3):244–53.
Aquino-Martinez R, Eckhardt BA, Rowsey JL, Fraser DG, Khosla S, Farr JN, et al. Senescent cells exacerbate chronic inflammation and contribute to periodontal disease progression in old mice. J Periodontol. 2021;92(10):1483–95.
Dong Y, Zhou H, Alhaskawi A, Wang Z, Lai J, Abdullah Ezzi SH, et al. Alterations in bone fracture healing associated with TNFRSF signaling pathways. Front Pharmacol. 2022;13: 905535.
Jeong E, Choi HK, Park JH, Lee SY. STAC2 negatively regulates osteoclast formation by targeting the RANK signaling complex. Cell Death Differ. 2018;25(8):1364–74.
Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2012;8(11):656–64.
Wahl EC, Aronson J, Liu L, Fowlkes JL, Thrailkill KM, Bunn RC, et al. Restoration of regenerative osteoblastogenesis in aged mice: modulation of TNF. J Bone Miner Res. 2010;25(1):114–23.
Lim JC, Ko KI, Mattos M, Fang M, Zhang C, Feinberg D, et al. TNFalpha contributes to diabetes impaired angiogenesis in fracture healing. Bone. 2017;99:26–38.
Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci USA. 2011;108(4):1585–90.
Dubon MJ, Yu J, Choi S, Park KS. Transforming growth factor beta induces bone marrow mesenchymal stem cell migration via noncanonical signals and N-cadherin. J Cell Physiol. 2018;233(1):201–13.
Blumenfeld I, Srouji S, Lanir Y, Laufer D, Livne E. Enhancement of bone defect healing in old rats by TGF-beta and IGF-1. Exp Gerontol. 2002;37(4):553–65.
Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A, et al. TGFbeta-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun. 2019;10(1):2795.
Xu J, Liu J, Gan Y, Dai K, Zhao J, Huang M, et al. High-dose TGF-beta1 impairs mesenchymal stem cell-mediated bone regeneration via Bmp2 inhibition. J Bone Miner Res. 2020;35(1):167–80.
Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823.
Kumar G, Roger PM. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. Int J Mol Sci. 2019;20(20):5154.
Karst M, Gorny G, Galvin RJ, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol. 2004;200(1):99–106.
Yao Z, Getting SJ, Locke IC. Regulation of TNF-induced osteoclast differentiation. Cells. 2021. https://doi.org/10.3390/cells11010132.
Xu Q, Ma H, Chang H, Feng Z, Zhang C, Yang X. The interaction of interleukin-8 and PTEN inactivation promotes the malignant progression of head and neck squamous cell carcinoma via the STAT3 pathway. Cell Death Dis. 2020;11(5):405.
Ye F, Li J, Xu P, Xie Z, Zheng G, Liu W, et al. Osteogenic differentiation of mesenchymal stem cells promotes c-Jun-dependent secretion of interleukin 8 and mediates the migration and differentiation of CD4(+) T cells. Stem Cell Res Ther. 2022;13(1):58.
Pratsinis H, Mavrogonatou E, Kletsas D. Scarless wound healing: from development to senescence. Adv Drug Deliv Rev. 2019;146:325–43.
Wilkinson HN, Hardman MJ. Senescence in wound repair: emerging strategies to target chronic healing wounds. Front Cell Dev Biol. 2020;8:773.
Maruyama M, Rhee C, Utsunomiya T, Zhang N, Ueno M, Yao Z, et al. Modulation of the inflammatory response and bone healing. Front Endocrinol. 2020;11:386.
Raffaele M, Vinciguerra M. The costs and benefits of senotherapeutics for human health. Lancet Healthy Longev. 2022;3(1):e67–77.
Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging. 2017;9(3):955–63.
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1):132–47.
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–58.
Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8(1):422.
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–17.
Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23(9):1072–9.
Sharma AK, Roberts RL, Benson RD Jr, Pierce JL, Yu K, Hamrick MW, et al. The senolytic drug navitoclax (ABT-263) causes trabecular bone loss and impaired osteoprogenitor function in aged mice. Front Cell Dev Biol. 2020;8:354.
Cleary JM, Lima CM, Hurwitz HI, Montero AJ, Franklin C, Yang J, et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest New Drugs. 2014;32(5):937–45.
He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000–11.
Hou JG, Jeon BM, Yun YJ, Cui CH, Kim SC. Ginsenoside Rh2 ameliorates doxorubicin-induced senescence bystander effect in breast carcinoma cell MDA-MB-231 and normal epithelial cell MCF-10A. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20051244.
Jin J, Lin J, Xu A, Lou J, Qian C, Li X, et al. CCL2: an important mediator between tumor cells and host cells in tumor microenvironment. Front Oncol. 2021;11: 722916.
Singh SK, Mishra MK, Eltoum IA, Bae S, Lillard JW Jr, Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep. 2018;8(1):1323.
Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front Immunol. 2019;10:877.
Kulkarni T, Kurundkar AR, Kim YI, de Andrade J, Luckhardt T, Thannickal VJ. The senescence-associated matricellular protein CCN1 in plasma of human subjects with idiopathic pulmonary fibrosis. Respir Med. 2020;161: 105821.
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–30.
Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016;23(6):188–98.
Rohn F, Kordes C, Buschmann T, Reichert D, Wammers M, Poschmann G, et al. Impaired integrin alpha(5) /beta(1) -mediated hepatocyte growth factor release by stellate cells of the aged liver. Aging Cell. 2020;19(4): e13131.
Thomas R, Wang W, Su DM. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing. 2020;17:2.
Morimura S, Takahashi K. Rac1 and stathmin but Not EB1 are required for invasion of breast cancer cells in response to IGF-I. Int J Cell Biol. 2011;2011: 615912.
Ikemoto-Uezumi M, Uezumi A, Tsuchida K, Fukada S, Yamamoto H, Yamamoto N, et al. Pro-insulin-like growth factor-ii ameliorates age-related inefficient regenerative response by orchestrating self-reinforcement mechanism of muscle regeneration. Stem Cells. 2015;33(8):2456–68.
Xu L, Wang Y, Wang J, Zhai J, Ren L, Zhu G. Radiation-induced osteocyte senescence alters bone marrow mesenchymal stem cell differentiation potential via paracrine signaling. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22179323.
Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G, et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4(11): e911.
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–14.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–31.
Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106(40):17031–6.
Wang T, He C. TNF-alpha and IL-6: the link between immune and bone system. Curr Drug Targets. 2020;21(3):213–27.
Hopkins B, Fisher J, Chang M, Tang X, Du Z, Kelly WJ, et al. An in-vitro study of the expansion and transcriptomics of CD(4+) and CD(8+) naive and memory T cells stimulated by IL-2, IL-7 and IL-15. Cells. 2022. https://doi.org/10.3390/cells11101701.
Wu Y, Tang Y, Zhang X, Chu Z, Liu Y, Tang C. MMP-1 promotes osteogenic differentiation of human bone marrow mesenchymal stem cells via the JNK and ERK pathway. Int J Biochem Cell Biol. 2020;129: 105880.
Zhang S, Wan Z, Pavlou G, Zhong AX, Xu L, Kamm RD. Interstitial flow promotes the formation of functional microvascular networks in vitro through upregulation of matrix metalloproteinase-2. Adv Funct Mater. 2022. https://doi.org/10.1002/adfm.202206767.
Niwa H, Kanno Y, Shu E, Seishima M. Decrease in matrix metalloproteinase-3 activity in systemic sclerosis fibroblasts causes alpha2-antiplasmin and extracellular matrix deposition, and contributes to fibrosis development. Mol Med Rep. 2020;22(4):3001–7.
Hilliard A, Mendonca P, Russell TD, Soliman KFA. The protective effects of flavonoids in cataract formation through the activation of Nrf2 and the inhibition of MMP-9. Nutrients. 2020. https://doi.org/10.3390/nu12123651.
Gao C, Ning B, Sang C, Zhang Y. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging (Albany NY). 2018;10(1):131–43.
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–33.
Saul D, Khosla S. Fracture healing in the setting of endocrine diseases, aging, and cellular senescence. Endocr Rev. 2022;43(6):984–1002.
Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586.
Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687–98.
Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. 2016;7:78–87.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Nos. 82172484, 81972523).
Author information
Authors and Affiliations
Contributions
ZQ, SC, and HL contributed to the study conception and design. KM, HT, and BM completed the literature collection. The first draft of the manuscript was written by SZ and ZQ. The manuscript was revised by RP, HCP, and HL. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, S., Qiao, Z., Pfeifer, R. et al. Modulation of fracture healing by senescence-associated secretory phenotype (SASP): a narrative review of the current literature. Eur J Med Res 29, 38 (2024). https://doi.org/10.1186/s40001-023-01604-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01604-7