
Abstract
Background
The phenotype of Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) patients is determined by the type of DMD gene variation, its location, effect on reading frame, and its size. The primary objective of this investigation was to determine the frequency and distribution of DMD gene variants (deletions/duplications) in Sri Lanka through the utilization of a combined approach involving multiplex polymerase chain reaction (mPCR) followed by Multiplex Ligation Dependent Probe Amplification (MLPA) and compare to the international literature. The current consensus is that MLPA is a labor efficient yet expensive technique for identifying deletions and duplications in the DMD gene.
Methodology
Genetic analysis was performed in a cohort of 236 clinically suspected pediatric and adult myopathy patients in Sri Lanka, using mPCR and MLPA. A comparative analysis was conducted between our findings and literature data.
Results
In the entire patient cohort (n = 236), mPCR solely was able to identify deletions in the DMD gene in 131/236 patients (DMD-120, BMD-11). In the same cohort, MLPA confirmed deletions in 149/236 patients [DMD-138, BMD -11]. These findings suggest that mPCR has a detection rate of 95% (131/138) among all patients who received a diagnosis. The distal and proximal deletion hotspots for DMD were exons 45–55 and 6–15. Exon 45–60 identified as a novel in-frame variation hotspot. Exon 45–59 was a hotspot for BMD deletions. Comparisons with the international literature show significant variations observed in deletion and duplication frequencies in DMD gene across different populations.
Conclusion
DMD gene deletions and duplications are concentrated in exons 45–55 and 2–20 respectively, which match global variation hotspots. Disparities in deletion and duplication frequencies were observed when comparing our data to other Asian and Western populations. Identified a 95% deletion detection rate for mPCR, making it a viable initial molecular diagnostic approach for low-resource countries where MLPA could be used to evaluate negative mPCR cases and cases with ambiguous mutation borders. Our findings may have important implications in the early identification of DMD with limited resources in Sri Lanka and to develop tailored molecular diagnostic algorithms that are regional and population specific and easily implemented in resource limited settings.
Similar content being viewed by others
Background
Duchenne muscular dystrophy (DMD), OMIM #310,200 and Becker muscular dystrophy (BMD), OMIM #300,376 are X-linked recessive disorders caused by pathogenic variations in the DMD gene (OMIM *300,377, HGNC ID: 29). These conditions are collectively referred to as dystrophinopathies [1, 2]. The prevalence of DMD and BMD, according to a recent meta-analysis, is 4.8 per 100,000 and 1.6 per 100,000, respectively [3]. It is important to note that mutations in the DMD gene predominantly affect males. However, there have been reports indicating that a percentage ranging from 2.5% to 7.8% of females have also been affected by these mutations, thereby being classified as symptomatic carriers. [4].
Receiving an accurate diagnosis of dystrophinopathy is crucial to avoid the lengthy and somber diagnostic odyssey. Even though the average age of diagnosis for DMD remained over a decade as 5 years, it has been reported that the average age of diagnosis in Europe has decreased below the age of 3 years, reflecting the impact of enhanced access to molecular diagnostics and increased primary physician awareness [5]. In contrast, diagnostic delays in DMD persist with notable frequency within traditionally marginalized populations encompassing individuals hailing from developing nations and those of a lower socioeconomic stratum [6, 7].
It can be challenging to differentiate DMD from BMD at a younger age. In this case, the "reading frame rule" [8] can help aid differential diagnosis, where DMD patients typically show out-of-frame deletions, whereas BMD patients typically show in-frame deletions [9]. The frame-shift hypothesis can predict the occurrence of DMD in 90% of cases and BMD in 94% of instances, with about 10% of genetic variations not adhering to the reading frame rule [10]. Exceptions to the reading frame rule highlight the intricacy of the condition and show that factors other than the reading frame affect how the dystrophin protein is expressed. These factors include the type of variation, where it is located within the DMD gene, and its size [11]. When evaluating the results of a molecular diagnosis to characterize dystrophinopathies, this cumulative impact of the type, size and localization of the variation is of importance.
To the best our knowledge, the present study is the first and the largest comprehensive genetic analysis of a cohort of 236 clinically suspected pediatric and adult myopathy cohort in a geographically defined South Asian population; Sri Lanka, using a combined approach of Multiplex PCR (mPCR) and Multiplex Ligation Dependent Probe Amplification (MLPA). The aims of this study are: (i) to determine the frequency and distribution of DMD gene variants (deletions/duplications) in Sri Lanka through the utilization of a combined approach involving mPCR followed by MLPA and compare to the international literature and (ii) to determine the applicability of the "reading frame rule" in Sri Lankan DMD/BMD patient population.
Methodology
Patient recruitment
A total of 236 patients [Age range (Mean); 1.5–42 Yrs (9 Yrs); Gender (Male-233:Female-3)] exhibiting characteristic clinical findings of Muscular Dystrophy were enrolled in the study from 2014 to 2022. Clinical diagnosis was based on the diagnostic recommendations by Bushby et al. [12]. Sociodemographic characteristics and clinical data of the patients were documented using a standard questionnaire and clinical batteries that included North Star Ambulatory Assessment (NSAA), Vignos Scale, Brook Scale and Medical Research Council Scale (MRC). Three females [age-9 Yrs (family history of elevated CPK; 9596 U/L, and NSAA-27/34), 10 Yrs (elevated CPK; 6786 U/L, NSAA- 23/34, no family history of symptoms) and 16 Yrs (elevated CPK; 3725 U/L, wheel chair bound at 15 Yrs of age and no family history of symptoms)], were too enrolled to assess the symptomatic carrier status.
Recruitment was conducted through neurology clinics in various government hospitals across Sri Lanka's Western, North-Western, North Central, Central, Southern, and Northern Provinces, as well as through pro bono mobile clinics and home visits. These patients were referred to the Interdisciplinary Center for Innovation in Biotechnology and Neuroscience (ICIBN) of the University of Sri Jayewardenepura until 2020, and then to the Institute for Combinatorial Advanced Research and Education (KDU-CARE), General Sir John Kotelawala Defence University (KDU), Sri Lanka for genetic testing.
Every participant provided written informed consent, where applicable. The assent of a proxy was obtained for patients unable to provide their own. This study adheres to the ethical standards of Sri Lankan institutional review boards that follow the Helsinki Declaration (Ethical Approval Nos. 449/09 and 38/19 from The Ethics Review Committee, Faculty of Medical Sciences, University of Sri Jayewardenepura, and Ethical Approval No. LRH/D/06/2007 Lady Ridgeway Hospital for Children, Sri Lanka).
Molecular Diagnostics
This study utilized the molecular diagnostic approach described in Wijekoon et al. [13] under the same corresponding author [13]. A summary of this approach is as follows. The initial diagnostic test for detecting deletions and duplications followed a level one testing approach, utilizing Multiplex PCR (mPCR) for 20 exons covering proximal and distal hot-spot regions of the DMD gene as described by Chamberlain et al. [14] and Beggs et al. [15] followed by the MLPA assay (MRC Holland SALSA MLPA Probe mixes P034 and P035) for all the clinically diagnosed dystrophinopathy patients. The diagnostic procedure was established utilizing the primary molecular diagnostic recommendations as outlined by Abbs et al. [17], as well as the revised edition by Fratter et al. [2], in alignment with the European Molecular Quality Genetics Network's (EMQN) optimal practice guidelines for genetic testing in dystrophinopathies [2, 16, 17]. To ascertain the impact of variations on the reading frame, the frame-shift checker available on the Leiden Muscular Dystrophy website (www.dmd.nl) was utilized to scrutinize all identified deletions and duplications where the number of patients who are following and not following the reading frame rule was identified. The comparative effectiveness of mPCR and MLPA was assessed by examining the individual capabilities of each method in detecting deletions and deletion boundaries of the DMD gene in genetically confirmed patients.
Comparative analysis with existing literature data from various countries representing diverse geographical regions
A literature review was conducted to compare the findings of this study, including the percentages of DMD gene deletion/duplication and the mean age of confirmatory molecular diagnosis, with existing literature data. The following method was employed for the literature review. The review process was structured into three primary stages: title screening, abstract screening, and document screening. A comprehensive search was conducted in globally recognized databases such as PubMed, Medline, Scopus, Embase, and Springer to identify relevant literature. The search was conducted using a combination of key words: Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, Mutation pattern, MLPA, and Diagnostic delay. A total of 861 publications were identified. All titles underwent a screening process, resulting in the selection of 275 documents for abstract screening. A total of 275 abstracts were reviewed, and 120 articles were identified as potentially meeting the inclusion criteria related to Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, MLPA, Mutation pattern, and Diagnostic delay. Ultimately, a comprehensive evaluation was conducted on the complete text of all 120 documents that were retained. This evaluation adhered to the same set of criteria for inclusion and exclusion as the initial screening of abstracts. As a result, a total of 49 papers were deemed suitable for inclusion in the subsequent comparative analysis.
Data analysis
Our findings on DMD gene variation types, hotspot locations, and deletion/duplication percentages, age at molecular diagnosis were compared with available data in literature from countries representing different geographical regions that have utilized the same molecular diagnostic protocol as our study. To test whether geographical region of various patient populations has an effect on the deletion/duplication percentages, the available country-specific data from the literature were graphically analyzed using boxplots followed by an ANOVA test. Those declared significantly different by ANOVA (p < 0.05) were then also studied using Tukey’s pairwise comparisons test. A comparative analysis was conducted to examine the mean age of confirmatory molecular diagnosis of DMD across countries representing Low and Middle-Income levels, as compared to countries representing High-Income levels. This analysis involved the use of boxplots to graphically represent the data. To ensure statistical power, the analysis categorized countries into two groups: Low and Middle-Income countries, and High-Income countries. This was necessary due to the limited availability of data on the mean age of confirmatory molecular diagnosis of DMD in only a few countries. Statistical analysis was performed using R Statistical software version 4.2.
Results
Demographic Characteristics of patient cohort
A total of 236 patients [Age range (Mean); 1.5–42 Yrs (9 Yrs); Gender (Male-233:Female-3)] exhibiting characteristic clinical findings of Muscular Dystrophy (Clinically diagnosed DMD-215, Clinically diagnosed BMD-21) were subjected to DMD gene deletion/ duplication analysis by mPCR and MLPA. Table 1 is a summary of demographic characteristics of the patient cohort.
Utility of mPCR and MLPA in the molecular diagnostics of the patient cohort
In the entire patient cohort (n = 236), mPCR solely was able to identify deletions in the DMD gene in 131/236 patients (DMD-120, BMD-11). In the same cohort, MLPA confirmed deletions in 149/236 patients [DMD-138, BMD -11]. Importantly deletion boundaries could be accurately detected by mPCR in a total of 100/236 (42%) patients. These findings suggest that mPCR has a detection rate of 95% (131/138) among all patients who received a diagnosis. Eighteen additional cases (18/236- 7.6%) (Detetions-5, Duplications-13) could be genetically diagnosed by MLPA over mPCR. The remaining 87 patients (37%) were negative for MLPA. Table 2 provides a summary of molecular diagnostic results achieved by mPCR and MLPA. Additional file 1: Table S1 summarizes the additional mutations and deletion borders identified by MLPA over mPCR (see Additional file 1: Table S1).
DMD gene deletions and duplications patterns in the Sri Lankan cohort
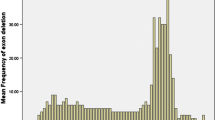
Table 2 provides a summary of the deletion and duplication variations, and their locations within the DMD gene. We observed clustering of deletion mutations in the exon 45–55 and 6–15 regions of the DMD gene and clustering of duplications in the exon 6–10 in our patient population (Fig. 1).

The deletion and duplication frequency by exons representing the clustering of deletion and duplication mutations in Sri Lankan dystrophinopathy patients. Clustering of deletion mutations in the exon 45–55 and 6–15 regions of the DMD gene and clustering of duplications in the exon 6–10. Red line represents DMD deletions, Blue line represents DMD duplications and the Green line represents BMD deletions
Comparative analysis of DMD gene deletion and duplication locations, percentages and the mean age of confirmatory molecular diagnosis, with existing literature data
We compared the variation hotspots of our cohort to the information available in the literature, as shown in Fig. 2. It was clear that South Asians represented a similar distal variation hotspot spanning exon 45–56 with the exception of the Netherlands, wherein the variations ranged from (exon 8–61). Exon 45–56 variation hotspot was consistent with the distal variation hotspots of the nations in South East Asia, East Asia, Europe, the USA–Canada, Latin America, the Middle East, and Africa. Furthermore, Peru, a country in Latin America, and Indonesia, a country in South East Asia, both had distinctive proximal hotspots ranging from (exon 18–30) and (exon 19–35), respectively. The proximal hotspots of Eastern European countries were notably spanning from (exon 45–49).

Comparative analysis of variability of DMD gene deletion and duplication hotspot location in different populations
Although it was reported that duplications could occur at random anywhere in the DMD gene, the comparative analysis allowed us to determine that in the majority of populations, duplications are concentrated in exons between (exon 2–20). For duplications ranging from (exon 50–79), (exon 42–55), and (exon 45–50), respectively, Iran from the Middle East, Taiwan from East Asia and, the African region stood out as unique clusters of duplications.
When the deletion and duplication percentages of studies from various geographical regions are compared, it was evident that the duplication percentages were significantly different (p < 0.05) among populations in South Asia Vs East Asia and South East Asia Vs East Asia. This is illustrated graphically in Fig. 3. The percentages of deletion, however, were not significantly different among populations in different geographical regions where South Asia Vs East Asia (p = 0.06) and South Asia Vs Europe (p = 0.06) were showing trends.

Data on country-specific percentages of duplications and deletions of the DMD gene from published literature were graphically evaluated for variability using boxplots
A comparative analysis was conducted on the mean age of confirmatory molecular diagnosis of DMD across countries representing Low and Middle-Income levels, namely Sri Lanka (our study), India [18], Thailand [19], Iran [20], Nepal [21], and Africa [22], versus to countries representing High-Income levels, including the USA [23], Eastern Europe, and Western Europe(5), based on the available literature. A significant difference (p = 0.001) was observed in the average age of confirmatory molecular diagnosis of DMD between Low and Middle-Income countries and High-Income Countries, as illustrated in Fig. 4.

The average age of confirmatory molecular diagnosis of DMD between Low and Middle-Income countries and High-Income countries, p = 0.001
The applicability of the "reading frame rule" in Sri Lankan DMD/BMD patient population
Upon determining the impact of variations in the DMD gene on the reading frame in our population, it was observed that 117/138 (84.7%) DMD cases were attributed to out of frame variations, while 17/138 (12.3%) exhibited in frame variations. The observed hotspot for in-frame variation for DMD within our population was identified as Exon 45–60. Interestingly, it was observed that 15/17 (88.2%) that did not adhere to the reading frame rule were associated with global developmental delay. This is described in Table 2.
Discussion
Based on our current understanding, this is the first and the largest study to use both mPCR and MLPA to conduct a genetic analysis on a cohort of 236 clinically suspected pediatric and adult myopathy patients in Sri Lanka (Table 1).
Utility of mPCR and MLPA in molecular diagnostics
The utilization of MLPA is presently regarded as a labor-efficient primary method for detecting deletions and duplications of single or multiple exons in the DMD gene, as approximately 70% of dystrophinopathy patients exhibit such genetic alterations [2]. Despite not being the primary molecular diagnostic method in developed nations, mPCR remains a viable and economical option for detecting deletions, and is, therefore, utilized in many laboratories situated in countries with limited resources [24]. In our study as represented in Table 2 and Supplementary Table 01, mPCR could provide molecular diagnosis for 55% (131/236) of the patients, of which 76% (100/136) of the patients the exact deletion boundaries accurately detected by mPCR. In line with our findings, studies conducted in South India [25] and North India [26] identified (103/150) 68% and (161/217) 74%, respectively, as the mutation detection Percentage by mPCR. Intriguingly in our study 95% (131/138) of the patients with deletion mutations could be diagnosed by mPCR. In line with our findings, Nouri et al., [27] identified a deletion detection rate of 95% for mPCR in an Iranian population.
On average, the cost of MLPA is estimated to be five times higher than that of mPCR. The utilization of MLPA as the principal screening technique within the framework of a developing nation would involve considerable costs. Hence, the proposed approach of employing mPCR as the primary step, followed by MLPA, is a prudent and precise method to efficiently proceed with the genetic diagnosis of DMD in settings with limited resources as previously described by Murugan et al. for South India [25].
Using MLPA diagnostics (Table 2), we were able to identify 82/138 (60%) patients as amenable to available exon skipping therapies. Interestingly, the majority of our patients were eligible for exon 51 skipping (30/82), followed by exon 53 skipping (19/82) and exon 45 skipping (19/82), which is consistent with data reported in the South Asian region, including North India [28], Tamil Nadu [29], and Pakistan[30]. The authors extensively discussed the identification of DMD patients who can benefit from exon skipping therapies in a previous study by Wijekoon et al. in 2023. Therefore, this paper does not extensively discuss this topic [13].
Table 3 presents our findings of newly identified DMD variants in our study population, which have not been previously documented in the Leiden Muscular Dystrophy Gene Variant Database. According to reports, the introduction of genetic material from distinct geographical populations has the potential to augment genetic diversity and potentially engender novel genotypic configurations within populations that are not isolated or indigenous [96]. The island of Sri Lanka, situated at the southernmost tip of South Asia and along the proposed Southern migration route, has been inhabited by diverse ethnic groups such as Portuguese, Dutch, British, and Arabs. This presents an interesting opportunity to gain a distinct perspective on the initial settlement of the subcontinent [97, 98]. In this context, we can hypothesize that the genetic admixtures that have taken place in Sri Lanka may have contributed to the emergence of the novel DMD variants that have been detected in our population.
Considering there have been reports of phenotypic differences in individuals carrying the same variation [11], healthcare practitioners should be more cautious when interpreting genomic results in the clinical environment for patients with similar variations and familial cases. Since distinct dystrophin-expressing tissues and cells may behave differently to specific defective dystrophins, such diversity is more common in BMD patients [33]. Furthermore, it is important to highlight that age can be confounded with phenotypic diversity when comparing patients with the same variation or familial cases. Therefore, it is advisable to take into account clinical batteries that are adjusted for age, such as the Weschler Intelligence Scale (WISC-IV). In this regard [99] have conducted a comprehensive analysis discussing the relationship between serum proteomics profile, cognitive assessment using WISC scores, and DMD gene mutations.
In this scenario, we have siblings in our patient cohort who have the same variation (out-of-frame deletion at exon 1–42), and whose Full-Scale IQ (FSIQ) as measured by the WISC-IV is 85 and 94, respectively. Additionally, one sibling with the identical variation was reported to have a motor development delay and scored FSIQ-85, but the other sibling did not (Table 3). Furthermore, the WISC-IV scores of another two distinct patients with identical variation (out-of-frame deletion at exon 45–52) were 83 and 67 for FSIQ, respectively. It's interesting that, despite the fact that this variation (deletion at exon 45–52) is likely to impact how brain dystrophin isoform Dp140 is expressed, only one patient outperformed the other on the WISC, indicating an intellectual deficit. These will serve as evidence of how complex it is to interpret genetic data in a clinical setting.
BMD patients with DMD variants in exon 1–8 and exon 41–45 impacting the Actin Binding Domain (ABD) and R16/R17 nNOS-binding domains are said to have a more severe presentation of BMD [1, 100]. We were unable to thoroughly evaluate this claimed connection due to the accumulation of DMD variants exclusively from exon 45–49 in our BMD cohort. Additionally, there have been reports on the comorbidity of Moyamoya disease [101], Frontometaphyseal Dysplasia [102], and Rippling muscle disease [103] with DMD. These findings highlight the intricacy of assessing the phenotypes of patients with dystrophinopathy in a clinical setting and underscore the need for the implementation of whole exome sequencing (WES) in the evaluation of dystrophinopathy patients exhibiting complex phenotypes and negative results on MLPA testing.
Deletion, duplication percentages and their location in the DMD gene
Accordingly as summarized in Table 2, 58% (136/236) of the cases in our sample were due to deletions as analyzed by MLPA [DMD- 90% (125/138) and BMD- 100% (11/11)], and 6% (13/236) of the cases (all DMD) were due to duplications. The DMD gene has a higher degree of allelic heterogeneity compared to many genes, due to the spontaneous mutation rate and large size, with 79 exons spanning 2.2 Mb, and hot spots for deletion mutations. One or more exons are deleted in 60–65% of DMD patients and 85% of BMD patients, respectively [8, 45].
Data from the literature demonstrate that deletion and duplication percentages vary across various populations. When deletion percentages for various ethnicities are taken into account (Fig. 3), East Asians; Japan (61%) [46], Taiwan (36%) [47], China (58% and 71%) [48, 49] and Korea (46% and 72%) [50, 51], demonstrate a trend of lower deletion percentages in the DMD gene compared to South Asians (p = 0.06); Sri Lanka (90%), pan India (73%-91%) [52], [34, 53, 54], and Pakistan (87%) [30] and European countries; Netherlands (63%) [55], Italy (65%) [56], Spain (71%) [38], Hungary (67%) [80], Poland (61%) [81], Russia 49% [82] and France (67%) [36]. However, Algeria; a Northern African county has a deletion percentage of 77% [57]. In contrary, a study by Selvaciti et al. [57] on an overall group of 258 patients from Eastern European countries (Bosnia, Bulgaria, Croatia, Hungary, Lithuania, Poland, Rumania, Serbia, Ukraine and Cyprus) identified a lower deletion percentage (27%) (Fig. 3).
Elhavary et al. [58] proposed that during Ancient Islamic times, Muslim immigration from the Levant and Africa, coupled with intermarriage, contributed to the reinforcement of gene flow of the DMD gene among the Saudi population. Turkey; a Middle Eastern country crossroads between Europe and Asia, has been found to exhibit a complex genetic makeup resulting from admixture with populations from the Balkans, Caucasus, Middle East, and Europe. Notably, genetic analyses have revealed a closer genetic affinity of the Turkish population to Europeans [59]. In this context, findings of two studies conducted by Cavdarli et al. [60] and Ulgenalp et al. [106] reported a deletion percentage of 92.4% and 63.7% in the Turkish population, respectively [60, 61]. These percentages were observed to be higher than the deletion percentage reported in the Saudi population, which was 46.3%. According to Elhavary et al. 2019, the higher percentage of deletions observed in Turkey compared to Saudi Arabia may be attributed to the admixture of Turkish populations with those of European descent [58]. However, a study conducted by Toksoy et al. 2019 on Turkish patients with DMD report a deletion percentage of 48.8%, which is similar to the percentage observed in the Saudi community [62]. Therefore, it is crucial to conduct further investigations on the hypothesis that European admixture results in higher deletion percentages. This can be achieved by studying larger patient cohorts, particularly those from South Asia (populations from the Indian Subcontinent) who were long-term subjects of Portuguese, Dutch, and British colonialism may provide unique resources to investigate.
However, it is important to note that the existing literature presents differing conclusions regarding the European admixture with the populations from Indian Subcontinent. According to the findings of Reich et al. in 2009, it was determined that the Ancestral North Indians (ANI) exhibit genetic similarities with individuals from the Middle East, Central Asia, and Europe [63]. Conversely, the "Ancestral South Indians" (ASI) were found to be genetically distinct from ANI. According to a study conducted by Neus Font-Porterias et al. in 2019, it was determined that the potential ancestral group of the proto-Roma, which is the largest transnational ethnic minority in Europe, can be traced back to a Punjabi group with minimal levels of West Eurasian ancestry [64]. Furthermore, the same study has revealed the presence of a multifaceted West Eurasian element, comprising approximately 65% of the Roma population. This finding can be attributed to the intermingling that transpired between non-proto-Roma groups and the Roma community during the period spanning from 1270 to 1580. Intriguingly, a recent study conducted by Perera et al. 2021 examined the four major ethnic groups in Sri Lanka, namely Sinhalese, Sri Lankan Tamils, Indian Tamils, and Moors. The study found that all Sri Lankan ethnicities, with the exception of Indian Tamils, exhibited a close clustering with populations from the Indian Bhil tribe, Bangladesh, and Europe. This clustering pattern suggests a shared Indo-Aryan ancestry among these populations [65].
Although consanguineous marriages are infrequent in Western societies, when Middle Eastern populations are considered; Iran (consanguinity: 50.7% in urban, 86.2% in rural) [66] Riyadh from Saudi Arabia (Consanguinity 80.6% in Samtah and 62.8% in Riyadh) [35] shows significant deletion percentages in DMD gene reported as 80% in Iran and 78% in Riyadh correspondingly. Elhavary et al. suggest that the observed higher consanguinity rate in Riyadh may have a link with the increased DMD deletion rates (77.8%) observed in Riyadh [58, 67]. Moreover, Algeria, a country in Northern Africa, has reported a higher rate of consanguinity (36.6%) [68]. Selvaciti et al. reported a noteworthy finding that Algerian patients exhibit a higher percentage (77%) of DMD deletions compared to Eastern Europeans [57], whose mutations are primarily nonsense (31%) followed by deletions (29%). Notably consanguinity account for 20–50% of marriages in various parts of Africa and Asia, particularly in South Asia [69]. In Pakistan and the southern portions of India, consanguineous marriages account for around 70% and 23% of all marriages, respectively [70]. In this context, it is possible to infer that consanguinity may have played a role in the higher rates of DMD gene deletions observed in South Asians, Africans, and Middle Eastern countries. However, it is important to note that while consanguineous unions can result in a higher occurrence of autosomal recessive disorders, there is ample evidence to suggest that consanguinity does not elevate the risk for autosomal dominant conditions or X-linked recessive conditions [71,72,73,74,75,76,77]. The available scientific evidence does not provide a strong basis for linking the higher rates of DMD gene deletions observed in South Asians, Africans, and Middle Eastern countries to consanguinity.
In this context, the increased frequencies of deletions in the DMD gene may be attributed to various mechanisms that are involved in the formation of genomic rearrangements [78,79,80]. Non-allelic homologous recombination (NAHR) is an important mechanism that can explain the frequencies of deletions and duplications [81,82,83]. The NAHR mechanism, specifically the crosslinking of Alu repeats, has been implicated as a causal factor in deletions affecting various genes, including the DMD gene [84]. However, it is important to note that if NAHR caused both deletions and duplications, it would anticipate comparable frequencies of deletions and duplications for each intron. However, this is not observed in the case of DMD [38]. Thereby it is reported that nonrecurrent events typically do not arise through NAHR. Instead, nonhomologous end joining (NHEJ), which involves the ligation of double-strand breaks, is often suggested as a mechanism for nonrecurrent intragenic deletions and duplications [85]. Several studies [86,87,88,89,90] have provided supporting evidence for this in DMD through the sequencing of deletion breakpoint junctions in the DMD gene. Moreover, it has also been proposed that duplications may occur at various stages of the cellular cycle. Similar to point mutations, deletions are primarily inherited from the maternal lineage, whereas duplications are passed down through the paternal germ line [91,92,93].
When the variation hotspots of our cohort were compared to the information available in the literature (Fig. 2), distinct distal hotspot has been identified for Netherlands, which ranged from (exon 8–61). This observed variation may have been influenced by clustering, which is connected to locally constrained gene flow across significant Dutch rivers and to country-wide ancestry gradients from neighboring territories [94]. For duplications ranging from (exon 50–79) and (exon 45–50), respectively, Iran from the Middle East, and, the African region stood out as unique hotspots. The observed uniqueness in duplication hotspots in Iran and Africa may be due to the high consanguinity rates associated with these populations.
The higher deletion frequency in the distal hotspot region (Fig. 1) and low duplication frequency observed in South Asians may provide insight into the feasibility of implementing conventional molecular diagnostic approaches such as mPCR, which can easily detect about 90% of the deletions in the hotspot region. Thus, it is proposed to develop tailored molecular diagnostic algorithms that are regional and population-specific and easily implemented in low resource settings. [95].
Delay in onset of the symptoms to molecular diagnosis
It is notable that diagnostic delays persist in traditionally disadvantaged groups, such as patients from developing countries and with lower socioeconomic status, because access to subspecialty care and genetic testing is difficult for patients from developing countries [7], including Sri Lanka [6]. (Fig. 4).
It is noteworthy that the average age of patients receiving their first clinical evaluation in our cohort was four years (Table 1), the same as the age of onset of symptoms. This is in contrast to data reported in India [18]; [Age at symptom onset—(3.7 ± 1.9 years), Age at first clinical evaluation – (8.1 ± 2.5 years)], China [104]; [Age at symptom onset- (3 years), Age at first clinical evaluation- (6–8 years)] Saudi Arabia[58]; [Age at symptom onset- (1–3 years), Age at first clinical evaluation- (9–12 years)]. Despite the fact that the average age of symptom onset in our patient cohort was 4 years (Table 1), when the first clinical examination was performed, only 21% of patients (29/138) were referred for molecular diagnostics before the age of 5 years. In our cohort, this has increased the average age of referral to molecular diagnostics to 7.8 years, indicating a delay in receiving an accurate diagnosis.
The observed delay may be attributable to the following: (1) The time required for referral to a specialist (Neurologist/ Pediatric Neurologist) and the difficulty in obtaining access to crucial diagnostic tests that must be performed at a government tertiary care hospital, (2) Lack of awareness among clinicians regarding the significance of molecular diagnostics as the gold standard for DMD confirmation; and (3) Neurogenetic testing is almost nonexistent in Sri Lankan government hospitals and only available at exorbitant costs in a few private sector centers. To the best of our knowledge, the neuromolecular diagnostic service established by the corresponding author at a government institute is the first of its kind to offer free molecular diagnostics for certain neuromuscular and neurodegenerative diseases. Moreover, CPK screening remains as the initial approach in testing for muscular dystrophies in resource limited settings where molecular diagnosis is not frequently available at a reduced cost. In our study, the majority of patients in our patient cohort underwent CPK evaluation during their initial visit to the healthcare professional (Table 1). This evaluation prompted their enrollment in the molecular diagnostic program, which provides genetic confirmation at no cost. The authors have previously addressed the relationship between age, mutation pattern and CPK levels in this patient cohort in a comprehensive manner, as documented in Wijekoon et al., 2023 [105]. Therefore, further discussion on this topic will not be included in the present paper. Thus CPK screening may be suggested in primary care as an approach in suspected early diagnosis of dystrophinopathy in resource limited settings which should be followed by a confirmatory molecular diagnostic approach, which will further reduce the diagnostic delay.
Despite being one of the most comprehensive studies conducted to date on dystrophinopathies in Sri Lanka, we acknowledge the following limitations in our study. N = 87 patients were negative for MLPA analysis; however, due to limited infrastructure and financial constraints, we could not perform genome sequencing for the MLPA negative cases, which we open up for future international collaboration. In addition, carrier detection for the mothers and female siblings of the probands was only conducted in a limited number of cases at the request of the consultant neurologist/pediatric neurologist due to the lack of genetic counselling services within the Sri Lankan health care system.
DMD gene variations interpretation from genetic report to clinic and the reading frame rule
It is important to remember that false positive results in MLPA can occur due to failed primer/probe binding, especially in single exon deletions. In this regard, according to Kim et al. 2016, MLPA has been found to have a false positive rate of approximately 15% in cases of large gene rearrangements that affect a single exon [31]. In addition, Buitrago and colleagues documented a 40% rate of false-positive results among individuals who were identified as having mutations affecting single exons through MLPA testing [32]. Hence, it is recommended that medical professionals in the clinic take into account the variation detection procedure utilized before drawing any conclusions about a single exon deletion [9]. The European Molecular Quality Genetics Network's (EMQN) best practice recommendations for genetic testing in dystrophinopathies include reconfirming single exon deletions discovered in MLPA by PCR [2]. In this analysis, we found n = 28 (20%) DMD patients with single exon deletions, with exon 44 and exon 51 being the most commonly deleted single exons (Table 2). Following EMQN protocols, all single exon deletions were re-confirmed by Multiplex PCR before being reported to the clinic.
To predict potential Duchenne or Becker effects of the deletion or duplication discovered using DNA data, the "reading frame rule" has gained popularity [8]. However, since deviations to the reading frame rules are frequent, the predictive sensitivity of the "reading frame rule" has been questioned. It can be difficult for medical practitioners to judge whether to classify a patient as having Duchenne or mild to moderate Becker by merely interpreting the reading frame, since "leaky" variations that are initially out-of-frame are found to produce low quantities of dystrophin, which will reduce the disease severity by 3–4% [33].
As presented in Table 2, in our population, a total of 17 out of 149 mutations, representing 11.4%, were found to be non-compliant with the reading frame rule. The comparative analysis of our value revealed that it is higher than the percentages reported in various regions including Tamil Nadu, India (3.9%) [29], Bangalore, India (8.4%)[34], Saudi Arabia (5.6%) [35], France (4%) [36], Italy (5.4%) [37], Brazil (9.6%) [38], TREAT-NMD DMD Global database (7%) [9], UMD-DMD database (4%) [36], and Leiden database (9%) [39]. However, our value is lower than the values reported in China (13.6%) [40] and Spain (15%) [41]. Mateu et al. [38] report that deletions exhibit a relatively low number of exceptions to the reading frame rule, whereas duplications and point mutations tend to have a greater probability of exceptions to the reading frame. In contrast, our cohort exhibited a reading frame exception rate of 8.7% for deletions and 2.6% for duplications. Nonetheless, the analysis of point mutations was not feasible in the present investigation, a constraint that we duly recognize.
When in-frame variations are evaluated further, it is reported in the literature that in-frame variations encoded by exons 64–70, 2–10, and 32–35 are associated with a DMD phenotype, as in-frame variations bordering the aforementioned regions will not produce a functional dystrophin protein [42]. Contrarily, 94% (16/17) of the in frame DMD patients in our dataset had a variational hotspot between exons 45 and 60 (Table 2). Consequently, it is suggested that the in-frame variational hotspot (exon 45–60) found in our study may represent a novel population-specific in-frame hotspot that has to be further studied in regional patient pools and validated via dystrophin protein levels.
In keeping with a previous study by Yan-Li Ma et al. 2022 [43] that revealed a predictive sensitivity of 86.8% for DMD, our cohort's predictive sensitivity for DMD based on the frame-shift theory was 85% (117/138) (Table 2). It is interesting to note that early gross motor development milestone delay is documented in the literature as a clinical feature of DMD but not BMD [44]. However, it is noteworthy that the gross motor development milestone delay, when taken alone has a limited ability to predict DMD, particularly in situations when the disease has in-frame variations [43].
Yan-Li Ma et al. 2022 [43] reported that, the reading-frame rule combined with the walking alone milestone significantly improved the early diagnosis rate of DMD, particularly the cases with in-frame variations, with a diagnostic coincidence rate increased to 93.49%, significantly higher than that predicted by reading-frame rule alone (P = 0.05). In this context, 15/17 (88%) of in-frame DMD cases in our study showed global development delay, of which, language delay accounting for 11/15 (73%) and motor development delay accounting for 13/15 (87%) of these cases, respectively (Table 2). It's interesting to note that none of the BMD patients in our dataset exhibit a generalized developmental delay (Table 1). Our findings thus provide more evidence in favor of the idea that the reading-frame rule should be combined with both language delay and motor development delay to increase the prediction sensitivity of DMD, particularly in situations when in-frame variations are present.
Conclusion
The largest and most well-established DMD mutation database in Sri Lanka demonstrates DMD gene deletions and duplications that are primarily concentrated in exons 45–55 and 2–20, respectively, which are consistent with the globally observed variation hotspots. Importantly, a unique, distinct mutation pattern of exon 45–60 was identified as a novel in-frame variation hotspot, which would contribute in personalized medicine to rational design of mutation-specific therapies. Furthermore, we have observed intriguing disparities in deletion and duplication frequencies when comparing our data to other Asian and Western populations.The utilization of mPCR as an initial molecular diagnostic method is considered highly feasible for countries with limited resources, owing to its 95% detection rate for deletions as identified in our study. Thereby, the authors propose an initial screening method using mPCR, then an assessment of cases that test negative for mPCR and have ambiguous mutation borders using MLPA. Our findings may have important implications in the early identification of DMD with limited resources in Sri Lanka and to develop tailored molecular diagnostic algorithms that are regional and population-specific and easily implemented in resource limited settings.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DMD:
-
Duchenne muscular dystrophy
- BMD:
-
Becker muscular dystrophy
- MLPA:
-
Multiplex Ligation Dependent Probe Amplification
- mPCR:
-
Multiplex PCR
- FSIQ:
-
Full Scale IQ
- WISC-IV:
-
Wechsler Intelligence Scale for Children
References
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJB, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135–44.
Fratter C, Dalgleish R, Allen SK, Santos R, Abbs S, Tuffery-Giraud S, et al. EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur J Hum Genet. 2020;28(9):1141–59.
Salari N, Fatahi B, Valipour E, Kazeminia M, Fatahian R, Kiaei A, et al. Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis. J Orthop Surg Res. 2022;17(1):1–12.
Lee SH, Lee JH, Lee K-A, Choi Y-C. Clinical and genetic characterization of female dystrophinopathy. J Clin Neurol. 2015;11(3):248–51.
Vry J, Gramsch K, Rodger S, Thompson R, Steffensen BF, Rahbek J, et al. European cross-sectional survey of current care practices for Duchenne muscular dystrophy reveals regional and age-dependent differences. Journal of neuromuscular diseases. 2016;3(4):517–27.
Samaranayake N, Dissanayaka P, Gunarathna I, Gonawala L, Wijekoon N, Rathnayake P, et al. What we fail to see in neuro-genetic diseases: a bird’s eye view from the developing world. Ann Neurosci. 2020;27(3–4):91–7.
Counterman KJ, Furlong P, Wang RT, Martin AS. Delays in diagnosis of Duchenne muscular dystrophy: an evaluation of genotypic and sociodemographic factors. Muscle Nerve. 2020;61(1):36–43.
Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–5.
Aartsma-Rus A, den Dunnen JT. Phenotype predictions for exon deletions/duplications: a user guide for professionals and clinicians using Becker and Duchenne muscular dystrophy as examples. Hum Mutat. 2019;40(10):1630–3.
Anwar S, He M, Lim KRQ, Maruyama R, Yokota T. A genotype-phenotype correlation study of exon skip-equivalent in-frame deletions and exon skip-amenable out-of-frame deletions across the DMD gene to simulate the effects of exon-skipping therapies: a meta-analysis. Journal of Personalized Medicine. 2021;11(1):46.
Gaina G, Vossen RH, Manole E, Plesca DA, Ionica E. Combining protein expression and molecular data improves mutation characterization of Dystrophinopathies. Front Neurol. 2021. https://doi.org/10.3389/fneur.2021.718396.
Birnkrant DJ, Bushby KM, Amin RS, Bach JR, Benditt JO, Eagle M, et al. The respiratory management of patients with Duchenne muscular dystrophy: a DMD care considerations working group specialty article. Pediatr Pulmonol. 2010;45(8):739–48.
Wijekoon N, Gonawala L, Ratnayake P, Sirisena D, Gunasekara H, Dissanayake A, et al. Gene therapy for selected neuromuscular and trinucleotide repeat disorders–An insight to subsume South Asia for multicenter clinical trials. IBRO Neuroscience Reports. 2023;14:146–53.
Chamberlain JS, Gibbs RA, Rainer JE, Nguyen PN, Thomas C. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16(23):11141–56.
Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86:45–8.
Wijekoon N, Gonawala L, Ratnayake P, Sirisena D, Gunasekara H, Dissanayake A, et al. Gene Therapy for selected Neuromuscular and Trinucleotide Repeat Disorders-An Insight to Subsume South Asia for Multicenter Clinical Trials. IBRO Neuroscience Reports. 2023. https://doi.org/10.1016/j.ibneur.2023.01.009.
Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR. Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul Disord. 2010;20(6):422–7.
Singh R-J, Manjunath M, Preethish-Kumar V, Polavarapu K, Vengalil S, Thomas PT, et al. Natural history of a cohort of Duchenne muscular dystrophy children seen between 1998 and 2014: an observational study from South India. Neurol India. 2018;66(1):77.
Yamputchong P, Pho-Iam T, Limwongse C, Wattanasirichaigoon D, Sanmaneechai O. Genotype and age at diagnosis in Thai boys with Duchenne muscular dystrophy (DMD). Neuromuscul Disord. 2020;30(10):839–44.
Zamani G, Hosseini Bereshneh A, Azizi Malamiri R, Bagheri S, Moradi K, Ashrafi MR, et al. The first comprehensive cohort of the Duchenne muscular dystrophy in Iranian population: mutation spectrum of 314 patients and identifying two novel nonsense mutations. J Mol Neurosci. 2020;70:1565–73.
Shrestha K, Shrestha S, Khatiwada S, Acharya B, Manandhar S, Pokharel RK. Multiplex ligation dependent probe amplification based mutation analysis of dystrophin gene in nepalese patients with duchenne muscular dystrophy. Nepal J Biotechnol. 2016;4(1):37–42.
Wonkam-Tingang E, Nguefack S, Esterhuizen AI, Chelo D, Wonkam A. DMD-related muscular dystrophy in Cameroon: clinical and genetic profiles. Mol Genet Genomic Med. 2020;8(8): e1362.
Thomas S, Conway KM, Fapo O, Street N, Mathews KD, Mann JR, et al. Time to diagnosis of Duchenne muscular dystrophy remains unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015. Muscle Nerve. 2022;66(2):193–7.
Aartsma-Rus A, Spitali P. Circulating biomarkers for Duchenne muscular dystrophy. J Neuromuscular Diseases. 2015;2(s2):S49–58.
Murugan SS, Chandramohan A, Lakshmi BR. Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis. Indian J Med Res. 2010;132(3):303–11.
Verma PK, Dalal A, Mittal B, Phadke SR. Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Human Genetics. 2012;18(1):91.
Nouri N, Fazel-Najafabadi E, Salehi M, Hosseinzadeh M, Behnam M, Ghazavi MR, et al. Evaluation of multiplex ligation-dependent probe amplification analysis versus multiplex polymerase chain reaction assays in the detection of dystrophin gene rearrangements in an Iranian population subset. Adv Biomed Res. 2014. https://doi.org/10.4103/2277-9175.125862.
Kohli S, Saxena R, Thomas E, Singh K, Bijarnia Mahay S, Puri RD, et al. Mutation spectrum of dystrophinopathies in India: implications for therapy. The Indian J Pediatrics. 2020;87:495–504.
Kumar SH, Athimoolam K, Suraj M, Das Christu Das MS, Muralidharan A, Jeyam D, et al. Comprehensive genetic analysis of 961 unrelated Duchenne Muscular Dystrophy patients: Focus on diagnosis, prevention and therapeutic possibilities. PLoS ONE. 2020;15(6):e0232654.
Ansar Z, Nasir A, Moatter T, Khan S, Kirmani S, Ibrahim S, et al. MLPA analyses reveal a spectrum of dystrophin gene deletions/duplications in Pakistani patients suspected of having Duchenne/Becker muscular dystrophy: a retrospective study. Genet Test Mol Biomarkers. 2019;23(7):468–72.
Kim MJ, Im Cho S, Chae J-H, Lim BC, Lee J-S, Lee SJ, et al. Pitfalls of multiple ligation-dependent probe amplifications in detecting DMD exon deletions or duplications. J Mol Diagn. 2016;18(2):253–9.
Buitrago T, García-Acero M, Guerra-Torres M, Pineda T, Gámez T, Suárez-Obando F, et al. Variants in the sequence of the probe hybridization site may affect MLPA performance in patients with Duchenne/Becker muscular dystrophy. J Appl Laboratory Med. 2023;8(3):469–78.
Hoffman EP. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myologica. 2020;39(4):179.
Polavarapu K, Preethish-Kumar V, Sekar D, Vengalil S, Nashi S, Mahajan NP, et al. Mutation pattern in 606 Duchenne muscular dystrophy children with a comparison between familial and non-familial forms: a study in an Indian large single-center cohort. J Neurol. 2019;266:2177–85.
El-Hazmi M, Al-Swailem A, Warsy A, Al-Swailem A, Sulaimani R, Al-Meshari A. Consanguinity among the Saudi Arabian population. J Med Genet. 1995;32(8):623–6.
Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype–phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD–DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6):934–45.
Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258:1610–23.
Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, Baena M, Verdura E, Nascimento A, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS ONE. 2015;10(8): e0135189.
White S, Den Dunnen J. Copy number variation in the genome; the human DMD gene as an example. Cytogenet Genome Res. 2006;115(3–4):240–6.
Yang J, Li SY, Li YQ, Cao JQ, Feng SW, Wang YY, et al. MLPA-based genotype–phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet. 2013;14:1–9.
Vieitez I, Gallano P, González-Quereda L, Borrego S, Marcos I, Millán J, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: Study of 284 cases. Neurología (English Edition). 2017;32(6):377–85.
Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145–51.
Ma YL, Zhang WH, Chen GH, Song LF, Wang Y, Yuan RL, et al. Walking alone milestone combined reading-frame rule improves early prediction of Duchenne muscular dystrophy. Front Pediatr. 2022. https://doi.org/10.3389/fped.2022.985878.
Mirski KT, Crawford TO. Motor and cognitive delay in Duchenne muscular dystrophy: implication for early diagnosis. J Pediatr. 2014;165(5):1008–10.
Koenig M, Hoffman E, Bertelson C, Monaco A, Feener C, Kunkel L. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17.
Okubo M, Goto K, Komaki H, Nakamura H, Mori-Yoshimura M, Hayashi YK, et al. Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J Rare Dis. 2017;12(1):1–7.
Hwa H-L, Chang Y-Y, Chen C-H, Kao Y-S, Jong Y-J, Chao M-C, et al. Multiplex ligation-dependent probe amplification identification of deletions and duplications of the Duchenne muscular dystrophy gene in Taiwanese subjects. J Formos Med Assoc. 2007;106(5):339–46.
Lin J, Li H, Liao Z, Wang L, Zhang C. Comparison of Carrier and de novo Pathogenic Variants in a Chinese DMD/BMD Cohort. Front Neurol. 2021;12:714677.
Kong X, Zhong X, Liu L, Cui S, Yang Y, Kong L. Genetic analysis of 1051 Chinese families with Duchenne/Becker muscular dystrophy. BMC Med Genet. 2019;20(1):1–6.
Suh MR, Lee K-A, Kim EY, Jung J, Choi WA, Kang S-W. Multiplex ligation-dependent probe amplification in X-linked recessive muscular dystrophy in Korean subjects. Yonsei Med J. 2017;58(3):613–8.
Lee BL, Nam SH, Lee JH, Ki CS, Lee M, Lee J. Genetic analysis of dystrophin gene for affected male and female carriers with Duchenne/Becker muscular dystrophy in Korea. J Korean Med Sci. 2012;27(3):274–80.
Tyagi R, Kumar S, Dalal A, Mohammed F, Mohanty M, Kaur P, et al. Repurposing pathogenic variants of DMD gene and its isoforms for DMD exon skipping intervention. Curr Genomics. 2019;20(7):519–30.
Vengalil S, Preethish-Kumar V, Polavarapu K, Mahadevappa M, Sekar D, Purushottam M, et al. Duchenne muscular dystrophy and Becker muscular dystrophy confirmed by multiplex ligation-dependent probe amplification: genotype-phenotype correlation in a large cohort. J Clin Neurol. 2017;13(1):91–7.
Deepha S, Vengalil S, Preethish-Kumar V, Polavarapu K, Nalini A, Gayathri N, et al. MLPA identification of dystrophin mutations and in silico evaluation of the predicted protein in dystrophinopathy cases from India. BMC Med Genet. 2017;18:1–10.
Van den Bergen J, Ginjaar H, Van Essen A, Pangalila R, De Groot I, Wijkstra P, et al. Forty-five years of Duchenne muscular dystrophy in the Netherlands. Journal of neuromuscular diseases. 2014;1(1):99–109.
Neri M, Rossi R, Trabanelli C, Mauro A, Selvatici R, Falzarano MS, et al. The genetic landscape of dystrophin mutations in Italy: a nationwide study. Front Genet. 2020;11:131.
Selvatici R, Rossi R, Fortunato F, Trabanelli C, Sifi Y, Margutti A, et al. Ethnicity-related DMD genotype landscapes in European and non-European countries. Neurol Genet. 2021. https://doi.org/10.1212/NXG.0000000000000536.
Elhawary NA, Jiffri EH, Jambi S, Mufti AH, Dannoun A, Kordi H, et al. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Hum Genomics. 2018;12(1):1–11.
Kars ME, Başak AN, Onat OE, Bilguvar K, Choi J, Itan Y, et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc Natl Acad Sci. 2021;118(36):e2026076118.
ÇAVDARLI B, YAYICI ÖK, CEYLAN AC, GÜNDÜZ CNS, TOPALOĞLU H. Genetic Landscape of Dystrofin Gene Deletions and Duplications From Turkey: A Single Center Experience. Türkiye Çocuk Hastalıkları Dergisi. 2021:1–6.
Karasoy H, Uran N, Dizdarer G, TtncoĞlu S, Dirik E. Deletion analysis and clinical correlations in patients with Xp21 linked muscular dystrophy. Turk J Pediatr. 2004;46:333–8.
Toksoy G, Durmus H, Aghayev A, Bagirova G, Rustemoglu BS, Basaran S, et al. Mutation spectrum of 260 dystrophinopathy patients from Turkey and important highlights for genetic counseling. Neuromuscul Disord. 2019;29(8):601–13.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461(7263):489–94.
Font-Porterias N, Arauna LR, Poveda A, Bianco E, Rebato E, Prata MJ, et al. European Roma groups show complex West Eurasian admixture footprints and a common South Asian genetic origin. PLoS Genet. 2019;15(9): e1008417.
Perera N, Galhena G, Ranawaka G. X-chromosomal STR based genetic polymorphisms and demographic history of Sri Lankan ethnicities and their relationship with global populations. Sci Rep. 2021;11(1):12748.
Saadat M, Zarghami M. Consanguineous marriages among Iranian Mandaeans living in south-west Iran. J Biosoc Sci. 2018;50(4):451–6.
Al-Jumah M, Majumdar R, Al-Rajeh S, Chaves-Carballo E, Salih MM, Awada A, et al. Deletion mutations in the dystrophin gene of Saudi patients with Duchenne and Becker muscular dystrophy. Saudi Med J. 2002;23(12):1478–82.
Chentouf A, Talhi R, Dahdouh A, Benbihi L, Benilha S, Oubaiche ML, et al. Consanguinity and epilepsy in Oran, Algeria: a case–control study. Epilepsy Res. 2015;111:10–7.
Mobarak AM, Chaudhry T, Brown J, Zelenska T, Khan MN, Chaudry S, et al. Estimating the health and socioeconomic effects of cousin marriage in South Asia. J Biosoc Sci. 2019;51(3):418–35.
Anwar S, Taslem Mourosi J, Arafat Y, Hosen MJ. Genetic and reproductive consequences of consanguineous marriage in Bangladesh. PLoS ONE. 2020;15(11):e0241610.
Temtamy S, Aglan M. Consanguinity and genetic disorders in Egypt. Middle East Journal of Medical Genetics. 2012;1(1):12–7.
Hamamy H. Consanguineous marriages: preconception consultation in primary health care settings. J Community Genet. 2012;3:185–92.
Hamamy HA, Masri AT, Al-Hadidy AM, Ajlouni KM. Consanguinity and genetic disorders. Saudi Med J. 2007;28(7):1015–7.
Shawky RM, Elsayed SM, Zaki ME, El-Din SMN, Kamal FM. Consanguinity and its relevance to clinical genetics. Egyptian J Med Human Genet. 2013. https://doi.org/10.1016/j.ejmhg.2013.01.002.
Woodcock IR, Fraser L, Norman P, Pysden K, Manning S, Childs AM. The prevalence of neuromuscular disease in the paediatric population in Yorkshire, UK; variation by ethnicity and deprivation status. Dev Med Child Neurol. 2016;58(8):877–83.
Landfeldt E. Consanguinity and autosomal recessive neuromuscular disorders. Dev Med Child Neurol. 2016;58(8):796–7.
Ben-Omran T, Al Ghanim K, Yavarna T, El Akoum M, Samara M, Chandra P, et al. Effects of consanguinity in a cohort of subjects with certain genetic disorders in Qatar. Mol Genet Genomic Med. 2020;8(1):e1051.
Hu X, Ray PN, Murphy EG, Thompson M, Worton R. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotypegenotype correlation. Am J Hum Genet. 1990;46(4):682.
Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet. 2000;34(1):297–329.
Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003;532(1–2):103–15.
Ji Y, Eichler EE, Schwartz S, Nicholls RD. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res. 2000;10(5):597–610.
Emanuel BS, Shaikh TH. Segmental duplications: an’expanding’role in genomic instability and disease. Nat Rev Genet. 2001;2(10):791–800.
Stankiewicz P, Lupski JR. Molecular-evolutionary mechanisms for genomic disorders. Curr Opin Genet Dev. 2002;12(3):312–9.
Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67(3):183–93.
Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283(1):1–5.
Ankala A, Kohn JN, Hegde A, Meka A, Ephrem CLH, Askree SH, et al. Aberrant firing of replication origins potentially explains intragenic nonrecurrent rearrangements within genes, including the human DMD gene. Genome Res. 2012;22(1):25–34.
Nobile C, Toffolatti L, Rizzi F, Simionati B, Nigro V, Cardazzo B, et al. Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum Genet. 2002;110:418–21.
Oshima J, Magner DB, Lee JA, Breman AM, Schmitt ES, White LD, et al. Regional genomic instability predisposes to complex dystrophin gene rearrangements. Hum Genet. 2009;126:411–23.
Baskin B, Stavropoulos DJ, Rebeiro PA, Orr J, Li M, Steele L, et al. Complex genomic rearrangements in the dystrophin gene due to replication-based mechanisms. Mol Genet Genomic Med. 2014;2(6):539–47.
Den Dunnen J, Grootscholten P, Bakker E, Blonden L, Ginjaar H, Wapenaar M, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. 1989;45(6):835.
Tuffery-Giraud S, Saquet C, Chambert S, Echenne B, Cuisset JM, Rivier F, et al. The role of muscle biopsy in analysis of the dystrophin gene in Duchenne muscular dystrophy: experience of a national referral centre. Neuromuscul Disord. 2004;14(10):650–8.
Grimm T, Meng G, Liechti-Gallati S, Bettecken T, Müller C, Müller B. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet. 1994;31(3):183–6.
Hu X, Ray PN, Worton RG. Mechanisms of tandem duplication in the Duchenne muscular dystrophy gene include both homologous and nonhomologous intrachromosomal recombination. EMBO J. 1991;10(9):2471–7.
Byrne RP, van Rheenen W, Consortium PMAG, van den Berg LH, Veldink JH, McLaughlin RL. Dutch population structure across space, time and GWAS design. Nature communications. 2020;11(1):4556.
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402.
Lavergne S, Molofsky J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc Natl Acad Sci. 2007;104(10):3883–8.
Thangaraj K, Chaubey G, Kivisild T, Reddy AG, Singh VK, Rasalkar AA, et al. Reconstructing the origin of Andaman Islanders. Science. 2005;308(5724):996-.
Mellars P, Gori KC, Carr M, Soares PA, Richards MB. Genetic and archaeological perspectives on the initial modern human colonization of southern Asia. Proc Natl Acad Sci. 2013;110(26):10699–704.
Wijekoon N, Gonawala L, Ratnayake P, Dissanayaka P, Gunarathne I, Amaratunga D, et al. Integrated genomic, proteomic and cognitive assessment in Duchenne Muscular Dystrophy suggest astrocyte centric pathology. Heliyon. 2023. https://doi.org/10.1016/j.heliyon.2023.e18530.
Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. 2016;87(4):401–9.
Park J, Jang W, Han JY. Differing disease phenotypes of Duchenne muscular dystrophy and Moyamoya disease in female siblings of a Korean family. Mol Genet Genomic Med. 2019;7(9):e862.
Kim J, Lee D-W, Jang J-H, Kim M, Yim J, Jang D-H. Case Report: Co-occurrence of Duchenne Muscular Dystrophy and Frontometaphyseal Dysplasia 1. Front Pediatr. 2021;9:628190.
Hiraide T, Ogata T, Watanabe S, Nakashima M, Fukuda T, Saitsu H. Coexistence of a CAV3 mutation and a DMD deletion in a family with complex muscular diseases. Brain Develop. 2019;41(5):474–9.
Wang D-N, Wang Z-Q, Yan L, He J, Lin M-T, Chen W-J, et al. Clinical and mutational characteristics of Duchenne muscular dystrophy patients based on a comprehensive database in South China. Neuromuscul Disord. 2017;27(8):715–22.
Wijekoon N, Gonawala L, Ratnayake P, Amaratunga D, Hathout Y, Mohan C, et al. Duchenne Muscular Dystrophy from Brain to Muscle: The Role of Brain Dystrophin Isoforms in Motor Functions. J Clin Med. 2023;12(17):5637.
Ulgenalp A, Giray O, Bora E, Hizli T, Kurul S, Sağin-Saylam G, Karasoy H, et al. Deletion analysis and clinical correlations in patients with Xp21 linked muscular dystrophy. Turk J Pediatr. 2004;46(4):333–8. https://pubmed.ncbi.nlm.nih.gov/15641267/.
Acknowledgements
We sincerely thank the patients and their family members for taking part in the study. Special thanks to the members of the Pro bono Research team of ICIBN of the University of Sri Jayewardenepura that included; Dr. Ruwani Wijayakoon, Dr. Vindika Suriyakumara, Dr. Chamara Jayasinghe, Dr. Beneeta Hettiarachchi, Dr. Roshani Karunarathne, Dr. Kasunka Gamage, Dr. Jayantha Udurawana, Dr. Navami Samaranayake, Dr. Gayatri Wijeweera, Dr. Kasinathar Lokajini, Dr. Pulasthi Dissanayaka Mr. Yoonus Imran, Ms. M.K.D.K. Attanayake and Ms. Shamali Wasala, for providing support. Moreover, we highly appreciate the technical support provided by Mr. Isuru Lokuge, a freelance analyst in performing statistical analysis.
Funding
The Corresponding author in Sri Lanka received funding from the Muscular Dystrophy Association Washington DC, USA (Grant Number FMS/7090/2010), The World Health Organization (WHO) (Grant Number 2010/81594-0), the World Class University Grant Project (University of Sri Jayewardenepura, Sri Lanka; Grant Numbers WCUP/Ph.D./19/2013 and WCUP/Ph.D./19B 2013), the Ministry of Primary Industries, Sri Lanka (Grant Number SP/CIN/2016/02, the University of Sri Jayewardenepura (Grant Numbers ASP/06/RE/2010/07, ASP/06/RE/2012/18, ASP/06/RE/2013/28), General Sir John Kotelawala Defence University, Sri Lanka (Grant Numbers KDU/RG/2021/CARE/005 and KDU/RG/2021/CARE/006), and the Interdisciplinary Center for Innovation in Biotechnology and Neuroscience, University of Sri Jayewardenepura (ICIBN/ USJ). Equipment was donated by the National Institutes of Health (Bethesda, MD, USA) through IBRO-APRC and by the Chinese Neuroscience Society. Moreover, the corresponding author has received funding from IBRO-APRC and the International Society for Neurochemistry (ISN) for international training scholarships for postgraduates and to conduct Neuroscience workshops in Sri Lanka.
Author information
Authors and Affiliations
Contributions
NW, and LG: Conceptualization; data curation; performed the experiments; analyzed and interpreted the data; wrote the original draft, reviewed and edited the paper to the final version. PR contributed in performing the clinical diagnosis and evaluation of myopathy patients. RL contributed reagents and consumables. DA contributed in statistical analysis, read and critically commented on the manuscript. YH read, critically commented on the manuscript. HS, AD, and EH are co-supervisors of NW and LG who provided academic guidance, read, critically commented on the manuscript. RDS: Conceptualization, supervision, project administration, analyzed and interpreted the data, contributed reagents and consumables, writing and reviewing the paper from its inception to the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study adheres to the ethical standards of Sri Lankan institutional review boards that follow the Helsinki Declaration (Ethical Approval Nos. 449/09 and 38/19, from The Ethics Review Committee, Faculty of Medical Sciences, University of Sri Jayewardenepura, and Ethical Approval No. LRH/D/06/2007 Lady Ridgeway Hospital for Children, Sri Lanka).
Consent for publication
Every participant provided written informed consent, where applicable. The assent of a proxy was obtained for patients unable to provide their own.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Additional mutations and deletion borders identified by MLPA over Multiplex PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wijekoon, N., Gonawala, L., Ratnayake, P. et al. Title-molecular diagnostics of dystrophinopathies in Sri Lanka towards phenotype predictions: an insight from a South Asian resource limited setting. Eur J Med Res 29, 37 (2024). https://doi.org/10.1186/s40001-023-01600-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01600-x