
Abstract
Key message
The range-wide level of genetic variation of Scots pine (Pinus sylvestris L.) is geographically structured. High admixture and low genetic structure of populations in Central Europe and Fennoscandia suggest past recolonization from multiple sources and the influence of human-mediated gene transfer. Gene pools of marginal and isolated stands require active conservation. Some areas of Scots pine distribution need further genetic studies.
Context
Scots pine (Pinus sylvestris L.) seems to be a species of low conservation priority because it has a very wide Eurasian distribution and plays a leading role in many forest tree breeding programs. Nevertheless, considering its economic value, long breeding history, range fragmentation, and increased mortality, which is also projected in the future, it requires a more detailed description of its genetic resources.
Aims
Our goal was to compare patterns of genetic variation found in biparentally inherited nuclear DNA with previous research carried out with mitochondrial and chloroplast DNA due to their different modes of transmission.
Methods
We analyzed the genetic variation and relationships of 60 populations across the distribution of Scots pine in Eurasia (1262 individuals) using a set of nuclear DNA markers.
Results
We confirmed the high genetic variation and low genetic differentiation of Scots pine spanning large geographical areas. Nevertheless, there was a clear division between European and Asian gene pools. The genetic variation of Asian populations was lower than in Europe. Spain, Turkey, and the Apennines constituted separate gene pools, the latter showing the lowest values of all genetic variation parameters. The analyses showed that most populations experienced genetic bottlenecks in the distant past. Ongoing admixture was found in Fennoscandia.
Conclusions
Our results suggest a much simpler recolonization history of the Asian than European part of the Scots pine distribution, with migration from limited sources and possible founder effects. Eastern European stands seem to have descended from the Urals refugium. It appears that Central Europe and Fennoscandia share at least one glacial refugium in the Balkans and migrants from higher latitudes, as well as from south-eastern regions. The low genetic structure between Central Europe and Fennoscandia, along with their high genetic admixture, may result at least partially from past human activities related to the transfer of germplasm in the nineteenth and early twentieth centuries. In light of ongoing climate changes and projected range shifts of Scots pine, conservation strategies are especially needed for marginal and isolated stands of this species. Genetic research should also be complemented in parts of the species distribution that have thus far been poorly studied.
Similar content being viewed by others

1 Introduction
The unique characteristics of forest trees—their long-life spans, relatively late reproductive maturity, economically important traits controlled by many genes, and the fact that they are generally undomesticated with large effective population sizes—make genetic studies hard to carry out and interpret. However, due to the high ecological and economic importance of forests and the social services they provide, forest tree genetics has rapidly evolved throughout the last few decades. The genetic diversity of trees serves two important purposes: (i) ensuring the stability and sustainability of forest ecosystems (Schaberg et al. 2008) by maintaining pre-existing genetic variation subjected to natural selection (Savolainen et al. 2007); and (ii) being the source of variability used by humans during breeding practices and improvement programs to increase forest productivity and adaptability to rapidly changing environments (Hedrick 2001).
The level and patterns of genetic variation observed today in forest tree species are mainly the consequence of their migration and range shifts after the Last Glacial Maximum (LGM), which took place approximately 25,000–18,000 years ago (Petit et al. 2003). A lot of genetic studies have been carried out to verify and accompany findings based on palynological and macrofossil data with an aim to trace the location of glacial refugia and postglacial recolonization routes. Most pine species constitute a unique study system in this matter because their mitochondrial and chloroplast genomes are transmitted in different ways (maternally via seeds and paternally via pollen). Therefore, the level and patterns of genetic variation revealed by mitochondrial and chloroplast markers are different (Petit et al. 2005). Mitochondrial DNA (mtDNA) still did not allow a clear inference of recolonization routes for many pine species, though a study by Donnelly et al. (2017) provides a set of new markers for finer-scale population genetic studies in pines. Their usefulness has already been shown for phylogeographic inferences in Scots pine by Wachowiak et al. (2022). Chloroplast DNA (cpDNA) is far less useful for such purposes due to its hypervariability, long-distance migration of pollen, and possible homoplasy (Provan et al. 2001). The nuclear genome, on the other hand, shows greater variability due to recombination and higher mutation rates (Wolfe et al. 1987).
Here, we focused on Scots pine (Pinus sylvestris L.), one of the most important forest-forming tree species in the Northern Hemisphere, which has high ecological and economic value, as well as breeding significance. Scots pine is a pioneer species that grows on poor and sandy soils (Farjon 2005). It is a key component of forest ecosystems in temperate and boreal zones, both in lowlands and mountain areas, growing either in pure stands or in mixed forests with other conifers and broad-leaf species (Richardson 1998). The vast Eurasian distribution of Scots pine spans a ca. 8000-km-long area from substantial populations in Scotland and the Iberian Peninsula in the west (6°W) to very far eastern areas of Siberia (150°E). The species crosses several climatic zones, starting from southern Spain and Turkey (40°N) up to the Arctic Circle (70°N) (Richardson 1998; Farjon 2005). It forms continuous populations across Central Europe, Fennoscandia, and Siberia, but the distribution along the western and southern range boundaries is fragmented. The gradual decline of populations and the expansion to higher latitudes are expected along the southern limit of Scots pine, mostly due to drought and biotic stresses (Tóth et al. 2017b).
The genetic variation and postglacial recolonization history of Scots pine have been extensively studied with molecular markers by numerous research groups, both in Europe and Asia (e.g., Buchovska et al. 2013; Cheddadi et al. 2006; Dering et al. 2017; Naydenov et al. 2007; Pyhäjärvi et al. 2008; Sannikov et al. 2014; Semerikov et al. 2014, 2018; Sinclair et al. 1999; Wachowiak et al. 2011, 2022; Wójkiewicz and Wachowiak 2016). The results show a high level of genetic variation of Scots pine, which is accompanied by remarkably low genetic structure across most of its distribution, especially based on cpDNA marker data. This situation seems to result from the high dispersal ability of Scots pine via pollen, but also from the postglacial migration history of this species (Semerikov et al. 2014, 2018; Wachowiak et al. 2022b). Still, there are no studies spanning large areas of Scots pine distribution based on nuclear markers, making it impossible to compare the level and patterns of genetic variation assessed with markers having different modes of transmission. Phylogeographic research indicates that Scots pine in Europe most likely survived the LGM in southern refugia—in the Iberian, Italian, and Balkan Peninsulas, as well as in Turkey. Most of Western Europe is dominated by just a few mitotypes, one of which is shared with the Iberian Scots pine populations (Cheddadi et al. 2006; Pyhäjärvi et al. 2008; Dering et al. 2017; Wachowiak et al. 2022), whereas Central and Northern Europe seem to have been recolonized from multiple sources, including cryptic refugia located at higher latitudes, but mainly by populations originating in the Balkan Peninsula (Pyhäjärvi et al. 2008; Wójkiewicz and Wachowiak 2016; Dering et al. 2017; Wachowiak et al. 2022). For the Asian part of the Scots pine distribution, genetic variation seems to be lower than that in the European part of the range (Semerikov et al. 2014, 2018; Wachowiak et al. 2022). The hypothetical Pleistocene refugia include numerous locations along the border of Russia with Kazakhstan and Mongolia and as far east as the Amur region (Sannikov and Petrova 2012; Sannikov et al. 2014, 2020).
In this study, we investigated the genetic diversity of 60 Scots pine populations in Eurasia. We analyzed the level and geographic patterns of genetic variation using nuclear microsatellite markers. Our goal was to verify if nuclear markers show similar genetic variation patterns as mitochondrial and chloroplast markers used in previous research considering their different modes of transmission. We also aimed to assess the role of admixture of populations from distinct recolonization lineages in shaping the level and distribution of genetic variation in Scots pine, especially in Central Europe and Fennoscandia. Finally, we discuss which parts of Scots pine distribution require special attention in terms of conservation of their gene pools.
2 Materials and methods
2.1 Sampling and DNA extraction
We sampled 60 populations of Scots pine in Eurasia, yielding a total of 1262 trees (Appendix Table 4; Fig. 1). Our dataset comprised locations that have different postglacial histories, as shown in previous studies. Needles were sampled from randomly selected, standing trees. Fresh needles collected from all individuals were stored at – 20 °C until extraction. DNA was subsequently extracted from 50 to 70 mg of tissue ground in liquid nitrogen according to the CTAB protocol (Dumolin et al. 1995). The protocol was modified to remove RNA from the samples by adding the final incubation step with 5 µl of RNase A (10 mg/ml) at 60 °C for 30 min. The DNA concentration was measured with a BioPhotometer plus spectrophotometer (Eppendorf AG, Germany) and adjusted to 15–20 ng/µl.

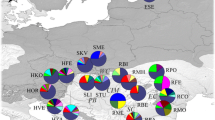
Genetic diversity maps of the analyzed Scots pine populations: IDW interpolation of A expected heterozygosity values (H E) and B allelic richness values (A R). Acronyms as in Appendix Table 4
2.2 Molecular analysis
All samples were genotyped with 19 nuclear microsatellite loci (Appendix Table 5). The selected markers were amplified in three multiplex PCRs using the Qiagen Multiplex PCR Kit (QIAGEN, Germany). A specific fluorescent dye was attached to the forward primer in each primer pair. The concentration of each primer was adjusted to yield even fluorescent signals. The multiplexes comprised the following markers: (I) psyl2, psyl16, psyl18, psyl25, psyl36, psyl42, psyl44, and psyl57; (II) spac11.4, spag7.14, ptTX2146, ptTX3025, ptTX4001, and ptTX4011; (III) spac11.8, spac12.5, pTctg4363, ptTX8446, and psyl17. Each multiplex PCR was performed in a total volume of 10 µl using the protocols described by Żukowska et al. (2017). The fluorescently labeled PCR products, along with the GeneScan™ 500 LIZ™ internal size standard (Thermo Fisher Scientific, USA), were separated on the Applied Biosystems® 3130xl Genetic Analyzer (Thermo Fisher Scientific, USA). The allele sizes were determined using GeneMapper™ software ver. 4.0 (Thermo Fisher Scientific, USA). The raw data were assessed manually and converted into discrete allele sizes using the automated binning software Tandem (Matschiner and Salzburger 2009). All size variants were also checked and adjusted manually.
2.3 Genetic variation and differentiation
To detect loci that deviated from Hardy–Weinberg equilibrium (HWE) in each population separately, we used the score test (U test) with a Markov chain (MC) algorithm to estimate the p values (Guo and Thompson 1992), implemented in the online version of GENEPOP ver. 4.7.5 (Rousset 2008). Both the heterozygote deficiency and heterozygote excess hypotheses were tested. In addition, we also employed the multisample version of the score test to check each locus across all populations. The same software was used to test linkage disequilibrium (LD) between each pair of loci in each population by using the log-likelihood ratio statistic (G-test) with the MC algorithm of Raymond and Rousset (1995). As we suspected the presence of null alleles in loci with significant deficiencies of heterozygotes, we checked the frequency of null alleles with a maximum likelihood method using the expectation–maximization (EM) algorithm of Dempster et al. (1977) available in FreeNA (Chapuis and Estoup 2007).
For each locus, population, and geographic region, we calculated basic genetic parameters, including the mean number of alleles (A), mean effective number of alleles (A E), number of private alleles (A P), and observed (H O) and expected heterozygosity (H E) using GenAlEx ver. 6.5 (Peakall and Smouse 2006, 2012). FSTAT ver. 2.9.4. (Goudet 2003) was used to compute the inbreeding coefficients (F IS) and mean rarefied allelic richness (A R). Subsequently, the values of H E and A R calculated for all populations were interpolated and presented on maps by using an inverse distance weighting (IDW) interpolation available in QGIS ver. 3.22 (QGIS Development Team 2021). In addition, we performed a linear regression analysis between the values of A, A E, A R, H O, H E, and the latitude of all populations using JMP® Pro ver. 16.1.0 (SAS Institute Inc. 2021). Considering the presence of null alleles, we also corrected the F IS values (F ISnull) with a Bayesian approach implemented in the INEst software ver. 2.2 (Chybicki and Burczyk 2009). The following parameters were set: 200,000 MC Monte Carlo cycles with every 20th cycle updated and a burn-in of 20,000. The deviance information criterion (DIC) was used to compare the full model (“nfb”—with null alleles, F IS > 0, and genotyping failures) with the random mating model (“nb”—with null alleles and genotyping failures; F IS = 0), thereby testing the significance of inbreeding.
2.4 Population structure
We assessed the genetic differentiation among the studied Scots pine populations grouped into geographic regions by computing pairwise fixation indexes (F ST) and pairwise Slatkin’s analogues of F ST (R ST) using the SPAGeDi ver. 1.5 computer package (Hardy and Vekemans 2002). A total of 10,000 permutations were performed to calculate the p values. Subsequently, principal coordinate analysis (PCoA), implemented in GenAlEx, was carried out to infer the genetic relationships among the geographic regions. Additionally, we used a Bayesian clustering approach implemented in BAPS ver. 6.0 (Corander et al. 2008) and STRUCTURE ver. 2.3.4 (Pritchard et al. 2000; Falush et al. 2003). Both programs perform well when the clusters are not well differentiated (F ST = 0.02–0.03), but they use different methods to search for the most likely number of clusters (Latch et al. 2006). For BAPS, we performed nonspatial genetic mixture clustering at the population level to define genetically divergent groups of populations. To this end, we initially tested all possible numbers of groups (K = 1–60) with 10 independent runs and repeated the analysis with 50 replicates for K = 1–12. Finally, we carried out admixture clustering using the obtained result of mixture clustering with parameters recommended by the authors. With STRUCTURE, we performed 500,000 iterations following a burn-in period of 50,000. We chose the admixture ancestry model with correlated allele frequencies. This configuration is considered best when the population structure is subtle (Falush et al. 2003). Twenty independent runs were set for K = 1–12. The output from STRUCTURE was visualized in the STRUCTURE HARVESTER web application (Earl and vonHoldt 2012). The optimum value of K was determined according to the Evanno method (Evanno et al. 2005). We aligned multiple runs for the optimum K value using the LargeKGreedy algorithm implemented in StructureSelector web-based software (Li and Liu 2018). StructureSelector was also used to generate STRUCTURE bar plots. Finally, to check the differentiation among populations, geographic regions, and the groups defined by the PCoA and Bayesian clustering, we ran the analysis of molecular variance (AMOVA) in Arlequin ver. 3.5.2.2 (Excoffier and Lischer 2010). A total of 10,000 permutations were performed to calculate the p values.
2.5 Phylogeographic structure, isolation by distance, and demographic history
We tested for a phylogeographic pattern among all populations, geographic regions, and populations within each geographic region using the permutation test by Hardy et al. (2003). Pairwise R ST and permuted R ST (pR ST) values were calculated and compared in SPAGeDi. P values were computed with 10,000 permutations. We subsequently checked whether differentiation among the Scots pine populations, as well as populations grouped into geographic regions, resulted from their isolation by distance (IBD) by performing a Mantel test implemented in GenAlEx. A total of 10,000 permutations were performed to calculate the p values. In the next step, we checked the possibility of bottlenecks, first by using the test developed by Cornuet and Luikart (1996) and implemented in INEst. This test is based on the excess of heterozygosity that can be observed as a consequence of a recent bottleneck compared to a demographically stable population (with constant size). As microsatellite markers do not mutate under a strict single-step mutation model (SMM), we chose a two-phase model (TPM) that incorporates both single-step and multistep mutations (Di Rienzo et al. 1994). The test was run with the parameters recommended by Peery et al. (2012). The number of coalescent simulations was set to 10,000. P values were determined with the Wilcoxon signed-rank test based on 1,000,000 permutations. We also used INEst to perform the test for the deficiency in M-Ratio (MR) (Garza and Williamson 2001), which is analogous to the test for the excess of heterozygosity but indicates bottlenecks that were more severe and took place in the distant past (Williamson-Natesan 2005). The simulation of a demographically stable population (MReq) was calculated as the mean of 10,000 coalescence replicates. The values of MR and MReq were compared for each population and the significance of MR < MReq was checked using the Wilcoxon signed-rank test based on 1,000,000 permutations.
We used an approximate Bayesian computation (ABC) framework implemented in DIYABC ver. 2.1.0 (Cornuet et al. 2014) to carry out demographic history modeling. We tested six demographic scenarios (Appendix Fig. 4). Populations were combined into four groups according to the results revealed by Bayesian clustering and PCoA: Spain/France (populations ES1-2, FR1-2); Fennoscandia/Central Europe/Balkans (NO1-2, SE, FI1-5, LT1-2, PL1-10, DE1-3, UA1-2, HU, RO, RS1-4, GR1-2); Eastern Europe (RU1-3); Urals/Asia (RU4-17). The old refugial Apennine and Turkish populations were excluded as outliers. Five scenarios were based on various versions of divergence events, whereas the sixth scenario assumed that populations from Eastern Europe result from admixture between European and Asian stands. Parameters used for computations are given in Appendix Table 6. Prior parameters for generation time, effective population sizes, the timing of events, and mutation rates were defined based on previous genetic and palynological studies. We performed 6 × 105 coalescent simulations for all scenarios. Simulations were repeated for the two best scenarios. For details on the best scenario choice methodology, please refer to Dering et al. (2021).
3 Results
3.1 Genetic polymorphism at the locus level
The final dataset consisted of 16 polymorphic loci for which the obtained electropherograms were of good quality, unambiguous, and reproducible (Żukowska et al. 2023b). Three markers were excluded due to inconclusive results (psyl16), insufficient quality (spag7.14), and no amplification in a large number of samples (spac11.8). The test for the presence of LD was significant in some populations for a few pairs of loci. Furthermore, some markers showed deviations from HWE. The mean frequency of null alleles averaged over all populations was Null = 0.018 (range, 0.001–0.088). Hence, the cut-off of Null = 0.19 (Chapuis et al. 2008), above which the value of H E is significantly underestimated, was not exceeded in any case. Inbreeding coefficients corrected for the presence of null alleles (F ISnull) were low for almost all loci, except psyl25, for which F ISnull = 0.285 (mean, 0.051). The results of the analysis using the INEst program showed that inbreeding was not a significant part of the model for ptTX4011, ptTX3025, and ptTX8446. In general, the markers from the “psyl” series were less polymorphic than the rest. A total of 242 alleles (A tot) were detected, resulting in a mean A tot = 15.13 per locus (range, 3–45). Among them, 38 were private. The mean number of alleles calculated across all populations was A = 5.675 (range, 1.267–16.200). Due to the presence of many alleles with low frequencies, the mean effective number of alleles across all populations was lower (A E = 3.298; range, 1.016–10.812). The mean rarefied allelic richness values were slightly higher for most loci (mean A R = 3.208; range, 1.059–6.313). The mean value of observed heterozygosity almost equalled the mean value of expected heterozygosity (H o = 0.526 and H E = 0.528). However, these values differed considerably for individual markers (Appendix Table 5).
3.2 Genetic variation and differentiation
The values of genetic variation parameters were generally high and geographically structured. The western parts of the Scots pine distribution (Scandinavia, Finland, Central Europe, Western Europe, the Balkans, and Spain) had the highest genetic variation, the eastern European parts and Turkey (Eastern Europe, Urals, and Turkey) had intermediate values, whereas the remaining Asian part had the lowest genetic variation (Western Siberia I, Western Siberia II, and Eastern Siberia) (Table 1). This western-eastern pattern was visible on the maps showing the IDW of H E and A R (Fig. 1). The Apennines were one exception, with the lowest values of all calculated parameters (A = 4.125, mean 8.212; A R = 3.902, mean 6.428; A E = 2.346, mean 3.595; H O = 0.426, mean 0.518; H E = 0.462, mean 0.532). The greatest variation was found among the values of A (mean, 8.212; range, 4.125–12.063), but it was due to the differences in the number of individuals from particular geographic regions, as the variation in the values of A R was much lower (mean, 6.428; range, 3.902–7.475) (Table 1). We found more private alleles (A P) in putative glacial refugial regions than in other parts of the Scots pine distribution (including Spain, the Italian Alps, Serbia, and southern part of the Urals) (Appendix Table 7). A linear regression analysis found no correlation between the values of genetic variation parameters and latitude (results not shown).
The genetic variation parameters were similar for individual populations within geographic regions, with few exceptions. Slightly lower values of the calculated genetic parameters were found for the Swedish population, one mountain and one peat-bog stand from Poland (PL6 and PL10, respectively), the Romanian population (RO, with only five individuals analyzed), and three locations in Asia (RU10, RU11, and RU13). Notably, the PL8 population from the Tatra Mountains had the highest values of A R = 3.600 (mean, 3.208), H O = 0.607 (mean, 0.526) and H E = 0.596 (mean, 0.528). The F ISnull values were very low in almost all stands (range, 0.004–0.061; mean, 0.029), except PL6 (F ISnull = 0.102) and PL10 (F ISnull = 0.443). Comparisons between the full model and the random mating model showed that inbreeding had a significant effect on the genetic variability of 26 out of 60 Scots pine populations (Appendix Table 7).
The level of interpopulation differentiation was low but significant (Table 2). The global value of the fixation index was F ST = 0.0276 (p < 0.001). The global value of the Slatkin’s analogue of F ST was higher (R ST = 0.0332; p < 0.001). Comparisons among geographic regions showed the distinctiveness of the Spanish and Apennine populations, with moderate pairwise values of R ST. The only exception was the greater similarity between Spain and Western Europe. Surprisingly, the R ST values between Eastern Siberia and Scandinavia, Finland, and Central Europe were not significant (p > 0.05). Scots pine from the Balkans was genetically closer to Scots pine from Finland and Central Europe than to the remaining geographic regions. In most cases, the values of F ST and R ST between adjacent geographic regions were low or nonsignificant (Table 2).
3.3 Population and phylogeographic structure, demographic history
The results of Bayesian clustering with BAPS and STRUCTURE were consistent. The most likely genetic structure identified with STRUCTURE consisted of two genetic groups (Fig. 2B): one comprising the Russian populations and the second including all other locations. The second most likely number of K was K = 5 (Fig. 2C), with the following genetic clusters: (1) Spain and France; (2) the Apennines; (3) Turkey; (4) the Urals and Asian part of Russia; and (5) the admixed group of the remaining populations. The analysis carried out in BAPS showed similar results but only three genetic clusters: (1) the Apennines; (2) the Urals and Asian part of Russia + one easternmost European population; and (3) the remaining European populations (Fig. 2D). The results of the Bayesian clustering with K = 5 were similar to the results of the PCoA (Fig. 3). The AMOVA outcomes confirmed that grouping into five genetic clusters best reflected the identified patterns of genetic differentiation, although the calculated values were low (F ST = 0.0228, R ST = 0.0371; p < 0.001) (Table 3).

Bayesian clustering of the Scots pine populations analyzed in the study. A Distribution of ΔK over K = 1–12 generated by STRUCTURE HARVESTER. B Bar plot showing STRUCTURE results for K = 2. C Bar plot showing STRUCTURE results for K = 5. D Bar plot showing BAPS results. The populations are separated by black vertical lines. The order of the populations corresponds to Appendix Table 4. Acronyms as in Table 1

PCoA among the studied Scots pine populations grouped into geographic regions
The comparison of the values of RST vs. pRST confirmed the existence of a phylogeographic structure among geographic regions (p = 0.0432) and populations (p = 0.0525). However, this is primarily the effect of marginal stands because at the level of geographic regions, the phylogeographic structure was significant only among populations within Western Europe (p = 0.0588), Western Siberia I (p < 0.001), and Western Siberia II (p = 0.0405). The Mantel test, although significant (p = 0.015), revealed almost no correlation between genetic and geographic distances for all populations (R2 = 0.018) (Appendix Fig. 5). The test for the excess of heterozygosity did not reveal any recent bottlenecks in any of the analyzed populations. Conversely, the calculated MRs were significantly below the mean MRseq for 41 out of 62 populations tested (Appendix Table 7). The only geographic region where none of the tests showed past bottlenecks was Spain.
The initial results of the ABC analysis showed that scenarios 3 and 4 are more likely than the others, although the estimated probability values were not high (21.07% and 20.93%, respectively). A re-analysis of these two best scenarios identified scenario 3 as the most likely (probability value 55.73%). The PCA for simulated data overlapped with the PCA results for the observed data (Appendix Fig. 6). According to the best scenario 3, populations from Eastern Europe diverged from the Urals/Asia group. The estimated divergence time was 1580 generations, i.e. (assuming a generation time of ~ 25 years) ~ 39,500 years BP. Divergence time back to the common ancestor was calculated to be ~ 4740 generations/118,500 years BP. The effective population size was the lowest for the Urals/Asia group (5180) and highest for the Fennoscandia/Central Europe/Balkans group (8790) (Appendix Table 8).
4 Discussion
Studies of genetic variation are particularly important in the era of climate change. Knowledge of neutral genetic variation, population history, and genetic relationships among different geographical regions is the first step of further research on the genetic basis of adaptive variation. To the best of our knowledge, this is the first comprehensive nuclear microsatellite-based study in the Eurasian range of Scots pine to address the question of admixture and homogenization of gene pools among populations of presumably distinct origins. Our results point to the differences between European and Asian Scots pine gene pools. We found that overall, Scots pine has high genetic variation, but this variation is significantly lower in its easternmost stands as well as in the Apennines and a few isolated areas. It also seems that the postglacial history of this species in Europe is very different from that in Asia, including recolonization from a higher number of glacial refugia, significant admixture, and the influence of human activities. Nuclear microsatellites seem to be useful in showing areas of secondary contact between once separated populations or the ones that have different origins.
4.1 The level and geographic pattern of genetic variation
Our results meet the expectations for a wind-pollinated tree species with a large census population size (Pyhäjärvi et al. 2020) and are in line with previous microsatellite and isoenzyme studies that showed substantial genetic variation but low among-population differentiation of Scots pine (Goncharenko et al. 1993; Prus-Głowacki et al. 1993; Provan et al. 1998; Bernhardsson et al. 2016; Wójkiewicz and Wachowiak 2016; Dering et al. 2017). We could distinguish the European and Asian gene pools, which differed from each other. These differences were evident both in the values of genetic variation parameters and in the Bayesian clustering, as well as in the PCoA. At the same time, the populations from Eastern Europe had a transitional character with intermediate genetic variation parameter values. This pattern definitely differs from the once widely accepted pattern of “southern richness to northern purity” (Hewitt 2000), which shows that Scots pine has a more complicated postglacial recolonization history.
Modes of transmission and mutation and migration rates differ for various marker types (mitochondrial, chloroplast, and nuclear) in pines. Thus, it is desirable to carry out phylogeographic studies with combined types of markers, especially since mtDNA, the only genome inherited in pines solely by seeds, evolves slowly and has a low level of intraspecific variation (Tóth et al. 2017b). The results of our study confirm the widely recognized locations of southern glacial refugia for Scots pine around the Mediterranean basin. In our study, we observed clear distinctiveness of the Apennine Peninsula. Likewise, Spanish populations also formed a separate genetic cluster. The pairwise FST/RST values between these two regions and the remaining areas were the highest. Among them, we detected less differentiation between Spain and Western Europe. Furthermore, Spanish and French stands grouped in the Bayesian clustering, showing a limited contribution of Iberian Scots pine populations to the recolonization of Western Europe. Demographic history modeling with DIYABC indicated that French and Spanish populations split from other parts of Europe already at the very beginning of the Last Glacial period (~ 118,500 years BP). Nevertheless, the best scenario (scenario 3) was only slightly more likely than scenario 4 which assumed that this divergence took place ~ 39,500 BP. Regardless of the time of divergence, the uniqueness of the gene pool of Iberian Scots pine, which we still observe for various types of genetic markers, confirms that the Pyrenees are a strong geographical barrier to gene flow (see also Sect. 4.3). According to our results, Turkish populations were only partly similar to Central and Northern European stands but were different from the eastern part of the Scots pine range, as evidenced by the PCoA, Bayesian clustering with STRUCTURE, and pairwise FST/RST. These results confirmed earlier findings of no or only little contribution of populations from the Iberian and Apennine Peninsulas, as well as from Turkey, to the northwards recolonization of Europe (Prus-Głowacki et al. 2003; Cheddadi et al. 2006; Pyhäjärvi et al. 2008; Wójkiewicz and Wachowiak 2016; Dering et al. 2017).
The origin of Scots pine in Central and Northern Europe is still a subject of much debate. The outcomes of this study support earlier reports indicating that Scots pine could migrate to Central Europe and far north from glacial refugia located in the Balkan Peninsula and in the Alps (e.g., Wachowiak et al. 2014; Wójkiewicz and Wachowiak 2016). In our research, populations from the Balkan Peninsula and Central and Northern Europe formed one genetic cluster with high admixture. Growing evidence of cryptic refugia at mid-latitudes makes it also possible that Scots pine in Central and Northern Europe descends from more refugial areas. The fact that we detected a high degree of admixture in Central and Northern Europe supports this hypothesis. Palynological and macrofossil data, as well as earlier studies with genetic markers, indicate the possible existence of northern, cryptic refugia of Scots pine in Central Europe in the Hungarian Plains, the Carpathians, and the Circum-Alpine area (e.g. Binney et al. 2009; Feurdean et al. 2011; Tóth et al. 2019), but also in northern latitudes in Scandinavia (Stewart et al. 2010). Unfortunately, our sampling was too limited to complete this picture, as we had only two populations from the Alps (CH and IT3), and just a few individuals from the Romanian Carpathians (RO). We also lack samples from the British Isles, where the origin of Scots pine is still a subject of debate (McGeever and Mitchell 2016; Sassoon et al. 2021).
Our study did not reveal the dual recolonization of Fennoscandia, which was shown in earlier works (Naydenov et al. 2007; Pyhäjärvi et al. 2008; Wachowiak et al. 2022). This result is particularly interesting, indicating that pollen-mediated gene transfer in areas of secondary contact may be efficient enough to blur a phylogeographic signal detected by mitochondrial markers transmitted by seeds. Therefore, nuclear microsatellites might be useful in showing areas of secondary contact between once separated populations or the ones that have different origins. This finding should be considered with caution as it may depend on the number and set of markers/populations used (Zimmer and Sønstebø 2018). The existence of the northeastern refugium somewhere in western Russia, hypothesized in several studies (Naydenov et al. 2007; Pyhäjärvi et al. 2008; Buchovska et al. 2013; Wachowiak et al. 2022), cannot be unequivocally confirmed or rejected based on our findings. Our demographic history modeling implied that it is more likely that Eastern European populations have descended not from the European refugium but rather from the Urals/Asian stands. It should be emphasized, however, that due to the high level of admixture, drawing more definitive conclusions would require the use of complex modeling and an increase in both the sampling geographical range as well as the number of trees from individual populations. A more detailed analysis of our results showed that populations from the Eastern European Plain (RU1-3) could in fact be divided into two groups (western: RU1-2 and eastern: RU3), the former being genetically more similar to Central and Northern Europe. It seems that the southern Urals refugium had a greater contribution to the genetic variation of RU3 than RU1-2, which is consistent with the findings of Vasilyeva et al. (2021). As the IBD was nonsignificant in this area, the observed pattern does not result from a geographic distance. These outcomes are in line with previous mtDNA studies that suggested a split between western and eastern populations of Scots pine approximately along the 38th meridian (Semerikov et al. 2018). Nevertheless, an unambiguous reconstruction of the demographic history of this region definitely requires exhaustive sampling, which takes into account also areas located in southwest Asia.
Our results point to the lower genetic variation of Scots pine in Asia and its rather uniform genetic structure. This is most likely the effect of a relatively simple recent demographic history of Scots pine in Asia, accompanied by limited admixture during expansion after the last glacial period. These results agree with previous findings based on mtDNA and cpDNA markers (Semerikov et al. 2014, 2018; Wachowiak et al. 2022). It should be mentioned that major parts of Russia, except the westernmost areas, remained ice-free during the LGM (Svendsen et al. 2004). For this reason, the number of glacial refugia was probably lower than in Europe. Moreover, gene flow could have had more time to homogenize Asian gene pools as compared to the European Scots pine. Our research indicates that this part of Scots pine distribution was recolonized by migrants from the southern Urals by a series of founder effects. Nevertheless, this conclusion needs further support with more thorough sampling from the Asian part of the Scots pine’s range. Apart from that, it seems that populations from the Urals have at least partially contributed not only to the contemporary variation in Eastern Europe but also, although to a lesser extent, to genetic variation in Central Europe and Finland. We cannot exclude the possibility that the area at the borders of Russia, Kazakhstan, and Mongolia served as another source of migrants. Nevertheless, our dataset comprised only several populations from Siberia and no samples from the far eastern parts. Unfortunately, palaeobotanical and genetic data on the Russian part of the Scots pine distribution are not sufficient to draw definite inferences (Sannikov et al. 2020). Therefore, more comprehensive studies are required to resolve the postglacial history of this region.
4.2 Genetic admixture and human-mediated gene transfer
When discussing admixture patterns, it is difficult to omit the recent history of Scots pine, characterized by intensive breeding that may have significantly affected original postglacial patterns and levels of genetic variation. As forests were overexploited during the medieval and early modern periods (Hosius et al. 2006), the reduction in forest cover eventually forced European countries to develop large-scale reforestation programs, leading to the increase in a forest area during the nineteenth and twentieth centuries (Mather 2001). In many cases, the remaining forests could not meet the high demand for seeds; thus, large amounts of germplasm, especially seeds, were transferred across Europe (Tulstrup 1959). It is usually impossible to trace back the historical transfer routes of forest reproductive material (FRM). Nevertheless, historical data about the time and geographical scales of FRM transfer are enough to conclude that they have undeniably affected the initial gene pools of Scots pine and other forest tree species. The trade and transfer of seeds of Scots pine, Norway spruce, European larch, and oaks started in the eighteenth century and continued throughout the nineteenth and early twentieth centuries (Tulstrup 1959; Jansen et al. 2017). Large-scale imports of non-native FRM to Fennoscandia began in the nineteenth century. During later afforestation efforts, non-autochthonous FRM of beech, oak, and Scots pine was widely introduced to Denmark, FRM of Norway spruce was transported to Sweden and Norway, and FRM of Scots pine was imported to Sweden (Myking et al. 2016). Most FRM was transferred from Germany, with little contribution of FRM from France, Belgium, and Hungary (Jansen et al. 2017). In contrast, Finland has limited records of FRM introductions (Myking et al. 2016). Reforestation efforts were not always successful. Hence, some countries tried to limit the use of foreign seeds and FRM from unknown sources. These failures eventually led to the establishment of the first international provenance trials and subsequent breeding programs. Today, the trade of FRM is regulated by European law and national regulations, but it is not obligatory to keep records about seed sources. Unfortunately, foresters and forest owners still tend to treat FRM as a cost that should be as low as possible. Furthermore, most Central and Northern European forests are intensely managed. Taking all these into account, almost no primary natural forests are left in Europe (Lorenz et al. 2005). Thus, virtually all genetic resources of Northern and Central European tree species have been shaped by a combination of natural processes, such as postglacial recolonization and local adaptation, and human impacts, including seed transfer, fragmentation, and silviculture (Ratnam et al. 2014; Żukowska et al. 2023a). Substantial admixture found in our study, especially in Central Europe and Fennoscandia, supports the view that the time and geographical scales of FRM transfer have undeniably affected the native gene pools of Scots pine.
4.3 Conservation implications
Genetic conservation of Scots pine seems to be of low priority because of its very wide distribution spanning several climatic zones. However, Scots pine in Europe has already experienced retreats from several sites in western and southern parts of continental Europe, as well as from the British Isles. These movements occurred due to a combination of climate change and anthropogenic disturbance, favoring the spread of broad-leaf species (Tóth et al. 2017b). Our research shows clear separation and a lower level of genetic variation of Scots pine from the Apennines than of populations occupying other geographic regions. It remains to be verified whether this is a matter of lower effective population sizes, range fragmentation, geographic isolation, or a combination of all these factors. Similar results were previously obtained by Belletti et al. (2012). The authors used nine nuclear microsatellites to analyze 20 populations of Scots pine from the Italian Alps and a single stand from the Apennines. The only population from the Apennines had the lowest values of genetic diversity parameters and constituted a separate genetic cluster. In another study, Scalfi et al. (2009) examined one Alpine and three Apennine Scots pine stands using three nuclear and two chloroplast microsatellites. The authors noticed the genetic distinctiveness of Apennine Scots pine and its possible origin from a separate glacial refugium. Nevertheless, they concluded that the studied Apennine populations still retain a high level of genetic diversity. As our study comprised two of the three nuclear microsatellites used by Scalfi et al. (2009), it emphasizes how the choice and number of markers can lead to distinct conclusions.
Pairwise comparisons of RST values between geographic regions in our study showed that, apart from the Apennines, also Spanish populations have a unique gene pool. RST indexes are supposed to be higher between populations that have been isolated from each other for a longer time (Slatkin 1995). Therefore, our study is another that confirms the long-lasting isolation and small contribution of Iberian populations to the recolonization of other parts of Europe due to the Pyrenees acting rather as a barrier than a bridge for gene exchange (Cheddadi et al. 2006; Pyhäjärvi et al. 2008; Dering et al. 2017; Prus-Głowacki et al. 1993; Wachowiak et al. 2022). Scots pine in the Iberian Peninsula inhabits disjunct mountainous areas with different climatic and ecological conditions. Individual populations are separated by large distances (Prus-Głowacki et al. 2003). Our study comprised only two stands from Spain but they are over 100 km apart with distinct microhabitats. Of these, population ES2 from Sierra de Gúdar is particularly interesting, as it is located at the eastern end of the Iberian System at an altitude of 2000 m a.s.l., which is usually covered with snow in winter, but it is also near the Mediterranean coast which makes the local climate milder than far inland. Both ES1 and ES2 had similar values of parameters connected with allelic diversity, but they slightly differed in heterozygosity values (HO = 0.555 vs. 0.519; HE = 0.567 vs. 0.516, respectively). It is also worth pointing out that none of the tests showed past bottlenecks, either in ES1 or ES2.
Geographically isolated populations are usually small and fragmented, making them more prone to genetic drift, inbreeding, and environmental changes. The study of the evolutionary potential of marginal populations is also important due to projected range shifts (Dyderski et al. 2018), which may lead to the loss of valuable gene pools. For Scots pine, many such populations occupy mountain ranges in southern peripheral areas. Studies from the Carpathian Mountains (Tóth et al. 2017a; Șofletea et al. 2020) showed that Scots pine growing in these territories has higher genetic variation than in non-mountainous areas, but the gene pools are not homogenous. They are also far less likely to be affected by the past human-mediated transfer of FRM. Recognition and conservation of the genetic pools in hard-to-reach and marginal areas should now be the priority. Whether the conservation policies should take the form of ex situ actions or rather of genetic rescue by promoting gene exchange, should be evaluated separately for each region. Unfortunately, our research comprised just a few isolated stands of Scots pine. We suppose that if we had more samples from the Carpathians and the Alps, the genetic variability of the European Scots pine range compared to its Asian part would be even higher. This would also allow more accurate inferences about human impact on the level and distribution of Scots pine’s genetic variation. In the context of comparing genetic variation between Europe and Asia, it would also be extremely important to thoroughly examine the Urals region.
5 Conclusion
This study provides important insights into the patterns of genetic variation of Scots pine across its Eurasian distribution. We revealed a high level of genetic admixture in Central and Northern Europe, which seems to be partially the result of postglacial recolonization from multiple glacial refugia but also human-mediated gene transfer. It appears that the Asian part of the Scots pine distribution has a different postglacial demographic history. The lower and homogenous genetic variation of Asian stands suggests their establishment by founder effects, possibly from the southern Urals refugium. This refugium had probably also served as the source of migrants that recolonized Eastern Europe, but this conclusion requires further research. Populations from the Apennines require special attention reflecting a general need for recognition and conservation of fragmented and marginal stands. Certainly, more comprehensive studies are necessary to unambiguously identify midlatitude hotspots of Scots pine genetic variation. In light of ongoing climate change, it is important to carry out genetic research in isolated, mountainous areas and those that thus far have been poorly covered: (1) Eastern Europe (Russia, Belarus, and Ukraine); (2) the southwestern Balkans (Bosnia and Herzegovina, Montenegro, Albania, and North Macedonia); (3) mid and high latitudes in Asia; and (4) the Russian Far East.
Availability of data and materials
The dataset analyzed during the current study is available in the Zenodo repository, https://zenodo.org/record/8405255.
References
Belletti P, Ferrazzini D, Piotti A, Moteleone I, Ducci F (2012) Genetic variation and divergence in Scots pine (Pinus sylvestris L.) within its natural range in Italy. Eur J Forest Res 131:1127–1138. https://doi.org/10.1007/s10342-011-0584-3
Bernhardsson C, Floran V, Ganea SL, García-Gil MR (2016) Present genetic structure is congruent with the common origin of distant Scots pine populations in its Romanian distribution. For Ecol Manage 361:131–143. https://doi.org/10.1016/j.foreco.2015.10.047
Binney HA, Willis KJ, Edwards ME, Bhagwat SA, Anderson PM, Andreev AA, Blaauw M, Damblon F, Haesaerts P, Kienast F, Kremenetski KV, Krivonogov SK, Lozhkin AV, MacDonald GM, Novenko EY, Oksanen P, Sapelko TV, Väliranta M, Vazhenina L (2009) The distribution of late-Quaternary woody taxa in northern Eurasia: evidence from a new macrofossil database. Quatern Sci Rev 28:2445–2464. https://doi.org/10.1016/j.quascirev.2009.04.016
Buchovska J, Danusevičius D, Baniulis D, Stanys V, Šikšnianienė J, KAvaliauskas D (2013) The location of the northern glacial refugium of Scots Pine based on mitochondrial DNA Markers. Balt for 19:2–12
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chapuis M-P, Lecoq M, Michalakis Y, Loiseau A, Sword GA, Piry S, Estoup A (2008) Do outbreaks affect genetic population structure? A worldwide survey in Locusta migratoria, a pest plagued by microsatellite null alleles. Mol Ecol 17:3640–3653. https://doi.org/10.1111/j.1365-294X.2008.03869.x
Cheddadi R, Vendramin GG, Litt T, François L, Kageyama M, Lorentz S, Laurent J-M, de Beaulieu J-L, Sadori L, Jost A, Lunt D (2006) Imprints of glacial refugia in the modern genetic diversity of Pinus sylvestris. Glob Ecol Biogeogr 15:271–282. https://doi.org/10.1111/j.1466-822X.2006.00226.x
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. https://doi.org/10.1093/jhered/esn088
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539. https://doi.org/10.1186/1471-2105-9-539
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014. https://doi.org/10.1093/genetics/144.4.2001
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc Ser B (Methodol) 39:1–38
Dering M, Kosiński P, Wyka TP, Pers-Kamczyc E, Boratyński A, Boratyńska K, Reich PB, Romo A, Zadworny M, Żytkowiak R, Oleksyn J (2017) Tertiary remnants and Holocene colonizers: genetic structure and phylogeography of Scots pine reveal higher genetic diversity in young boreal than in relict Mediterranean populations and a dual colonization of Fennoscandia. Divers Distrib 23:540–555. https://doi.org/10.1111/ddi.12546
Dering M, Baranowska M, Beridze B, Chybicki IJ, Danelia I, Iszkuło G, Kvartskhava G, Kosiński P, Rączka G, Thomas PA, Tomaszewski D, Walas Ł, Sękiewicz K (2021) The evolutionary heritage and ecological uniqueness of Scots pine in the Caucasus ecoregion is at risk of climate changes. Sci Rep 11:22845. https://doi.org/10.1038/s41598-021-02098-1
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci U S A 91:3166–3170. https://doi.org/10.1073/pnas.91.8.3166
Donnelly K, Cottrell J, Ennos RA, Vendramin GG, A’Hara S, King S, Perry A, Wachowiak W, Cavers S (2017) Reconstructing the plant mitochondrial genome for marker discovery: a case study using Pinus. Mol Ecol Resour 17:943–954. https://doi.org/10.1111/1755-0998.12646
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256. https://doi.org/10.1007/BF00220937
Dyderski MK, Paź S, Frelich LE, Jagodziński AM (2018) How much does climate change threaten European forest tree species distributions? Glob Change Biol 24:1150–1163. https://doi.org/10.1111/gcb.13925
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. https://doi.org/10.1093/genetics/164.4.1567
Farjon A (2005) Pines: drawings and descriptions of the genus Pinus, 2nd edn. Brill, Leiden
Feurdean A, Tanţău I, Fărcaş S (2011) Holocene variability in the range distribution and abundance of Pinus, Picea abies, and Quercus in Romania; implications for their current status. Quatern Sci Rev 30:3060–3075. https://doi.org/10.1016/j.quascirev.2011.07.005
Garza J, Williamson E (2001) Detecting the reduction in population size using data from microsatellite DNA. Mol Ecol 10:305–318. https://doi.org/10.1046/j.1365-294x.2001.01190.x
Goncharenko GG, Padutov VE, Silin AE (1993) Allozyme variation in natural populations of Eurasian pines. II. Genetic variation, diversity, differentiation and gene flow in Pinus sibirica Du Tour in some lowland and mountain populations. Silvae Genetica 42:246–253
Goudet J (2003) Fstat (ver. 2.9.4), a program to estimate and test population genetics parameters. Available from https://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 23 Feb 2023
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361. https://doi.org/10.2307/2532296
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8286.2002.00305.x
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hedrick PW (2001) Conservation genetics: Where are we now? Trends Ecol Evol 16:629–636. https://doi.org/10.1016/S0169-5347(01)02282-0
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. https://doi.org/10.1038/35016000
Hosius B, Leinemann L, Konnert M, Bergmann F (2006) Genetic aspects of forestry in the Central Europe. Eur J Forest Res 125:407–417. https://doi.org/10.1007/s10342-006-0136-4
Jansen S, Konrad H, Geburek T (2017) The extent of historic translocation of Norway spruce forest reproductive material in Europe. Ann for Sci 74:1–17. https://doi.org/10.1007/s13595-017-0644-z
Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE (2006) Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet 7:295–302. https://doi.org/10.1007/s10592-005-9098-1
Li Y-L, Liu J-X (2018) StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177. https://doi.org/10.1111/1755-0998.12719
Lorenz M, Fischer R, Mues V (2005) Forest resources in Europe and their condition. In: Geburek T, Turok J (eds) Conserving and management of forest genetic resources in Europe. Arbora Publishers, Slovakia, pp 111–126
Mather AS (2001) The transition from deforestation to reforestation in Europe. In: Angelsen A, Kaimowitz D (eds) Agricultural technologies and tropical deforestation. CABI in association with Centre of International Forestry Research, United Kingdom, p 35–52
Matschiner M, Salzburger W (2009) TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics 25:1982–1983. https://doi.org/10.1093/bioinformatics/btp303
McGeever AH, Mitchell FJG (2016) Re-defining the natural range of Scots Pine (Pinus sylvestris L.): a newly discovered microrefugium in western Ireland. J Biogeogr 43:2199–2208. https://doi.org/10.1111/jbi.12761
Myking T, Rusanen M, Steffenrem A, Kjær ED, Jansson G (2016) Historic transfer of forest reproductive material in the Nordic region: drivers, scale and implications. Forestry 89:325–337. https://doi.org/10.1093/forestry/cpw020
Naydenov K, Senneville S, Beaulieu J, Tremblay F, Bousquet J (2007) Glacial vicariance in Eurasia: mitochondrial DNA evidence from Scots pine for a complex heritage involving genetically distinct refugia at mid-northern latitudes and in Asia Minor. BMC Evol Biol 7:233. https://doi.org/10.1186/1471-2148-7-233
Peakall R, Smouse PE (2006) GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Petit RJ, Aguinagalde I, de Beaulieu J-L, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565. https://doi.org/10.1126/science.1083264
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005) INVITED REVIEW: comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701. https://doi.org/10.1111/j.1365-294X.2004.02410.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Provan J, Soranzo N, Wilson NJ, McNicol JW, Forrest GI, Cottrell J, Powell W (1998) Gene-pool variation in Caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple-sequence repeats. Proc Biol Sci 265:1697–1705. https://doi.org/10.1098/rspb.1998.0491
Provan J, Powell W, Hollingsworth PM (2001) Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol 16:142–147. https://doi.org/10.1016/S0169-5347(00)02097-8
Prus-Głowacki W, Urbaniak L, Zubrowska-Gil M (1993) Allozyme differentiation in some European populations of Scots pine (Pinus sylvestris L.). Genetica Polonica 34:159–176
Prus-Głowacki W, Stephan BR, Bujas E, Alia R, Marciniak A (2003) Genetic differentiation of autochthonous populations of Pinus sylvestris (Pinaceae) from the Iberian Peninsula. Plant Syst Evol 239:55–66. https://doi.org/10.1007/s00606-002-0256-3
Pyhäjärvi T, Salmela MJ, Savolainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4:247–254. https://doi.org/10.1007/s11295-007-0105-1
Pyhäjärvi T, Kujala ST, Savolainen O (2020) 275 years of forestry meets genomics in Pinus sylvestris. Evol Appl 13:11–30. https://doi.org/10.1111/eva.12809
QGIS Development Team (2021) QGIS geographic information system. Open Source Geospatial Foundation Project. Available from: http://qgis.osgeo.org. Accessed 23 Feb 2023
Ratnam W, Rajora OP, Finkeldey R, Aravanopoulos F, Bouvet J-M, Vaillancourt RE, Kanashiro M, Fady B, Tomita M, Vinson C (2014) Genetic effects of forest management practices: global synthesis and perspectives. For Ecolog Manag 333:52–65. https://doi.org/10.1016/j.foreco.2014.06.008
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283. https://doi.org/10.1111/j.1558-5646.1995.tb04456.x
Richardson DM (1998) Ecology and biogeography of Pinus. Cambridge University Press, Cambridge
Rousset F (2008) Genepop’007: a complete re-implementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Sannikov SN, Petrova IV (2012) Phylogenogeography and genotaxonomy of Pinus sylvestris L. populations. Russ J Ecol 43:273–280. https://doi.org/10.1134/S1067413612040145
Sannikov SN, Petrova IV, Egorov EV, Sannikova NS (2014) A system of Pleistocene refugia for Pinus sylvestris L. in the southern marginal part of the species range. Russ J Ecol 45:167–173. https://doi.org/10.1134/S1067413614030102
Sannikov SN, Petrova IV, Egorov EV, Sannikova NS (2020) Searching for and revealing the system of Pleistocene refugia for the species Pinus sylvestris L. Russ J Ecol 51:215–223. https://doi.org/10.1134/S1067413620030133
SAS Institute Inc. (2021) JMP®, Version 16.1.0. SAS Institute Inc., USA, 1989–2021
Sassoon D, Fletcher WJ, Hotchkiss A, Owen F, Feng L (2021) Scots pine (Pinus sylvestris) dynamics in the Welsh Marches during the mid to late-Holocene. Holocene 31:1033–1046. https://doi.org/10.1177/0959683621994654
Savolainen O, Pyhäjärvi T, Knürr T (2007) Gene flow and local adaptation in trees. Annu Rev Ecol Evol Syst 38:595–619. https://doi.org/10.1146/annurev.ecolsys.38.091206.095646
Scalfi M, Piotti A, Rossi M, Piovani P (2009) Genetic variability of Italian southern Scots pine (Pinus sylvestris L.) populations: the rear edge of the range. Eur J Forest Res 128:377–386. https://doi.org/10.1007/s10342-009-0273-7
Schaberg PG, DeHayes DH, Hawley GJ, Nijensohn SE (2008) Anthropogenic alterations of genetic diversity within tree populations: implications for forest ecosystem resilience. For Ecol Manage 256:855–862. https://doi.org/10.1016/j.foreco.2008.06.038
Semerikov VL, Semerikova SA, Dymshakova OS, Zatsepina KG, Tarakanov VV, Tikhonova IV, Ekart AK, Vidyakin AI, Jamiyansuren S, Rogovtsev RV, Kalchenko LI (2014) Microsatellite loci polymorphism of chloroplast DNA of Scots pine (Pinus sylvestris L.) in Asia and Eastern Europe. Russ J Genet 50:577–585. https://doi.org/10.1134/S1022795414040127
Semerikov VL, Semerikova SA, Putintseva YA, Tarakanov VV, Tikhonova IV, Vidyakin AI, Oreshkova NV, Krutovsky KV (2018) Colonization history of Scots pine in Eastern Europe and North Asia based on mitochondrial DNA variation. Tree Genet Genomes 14:8. https://doi.org/10.1007/s11295-017-1222-0
Sinclair WT, Morman JD, Ennos RA (1999) The postglacial history of Scots pine (Pinus sylvestris L.) in western Europe: evidence from mitochondrial DNA variation. Mol Ecol 8:83–88. https://doi.org/10.1046/j.1365-294X.1999.00527.x
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462. https://doi.org/10.1093/genetics/139.1.457
Șofletea N, Mihai G, Ciocîrlan E, Curtu AL (2020) Genetic diversity and spatial genetic structure in isolated Scots Pine (Pinus sylvestris L.) populations native to Eastern and Southern Carpathians. Forests 11:1047. https://doi.org/10.3390/f11101047
Stewart JR, Lister AM, Barnes I, Dalén L (2010) Refugia revisited: individualistic responses of species in space and time. Proc Biol Sci 277:661–671. https://doi.org/10.1098/rspb.2009.1272
Svendsen J, Alexanderson H, Astakhov V, Demidov I, Dowdeswell JA, Funder S, Gataullin V, Henriksen M, Hjort C, Houmark-Nielsen M, Hubberten HW, Ingólfsson Ó, Jakobsson M, Kjær KH, Larsen E, Lokrantz H, Lunkka JP, Lyså A, Mangerud J, Matiouchkov, Murray AA, Möller P, Niessen F, Nikolskaya O, Polyak L, Saarnisto M, Siegert C, Siegert MJ, Spielhagen RF, Stein R (2004) Late Quaternary ice sheet history of Northern Eurasia. Quat Sci Rev 23: 1229-1271. https://doi.org/10.1016/j.quascirev.2003.12.008
Tóth EG, Vendramin GG, Bagnoli F, Cseke K, Höhn M (2017a) High genetic diversity and distinct origin of recently fragmented Scots pine (Pinus sylvestris L.) populations along the Carpathians and the Pannonian Basin. Tree Genet Genomes 13:47. https://doi.org/10.1007/s11295-017-1137-9
Tóth EG, Köbölkuti ZA, Pedryc A, Höhn M (2017b) Evolutionary history and phylogeography of Scots pine (Pinus sylvestris L.) in Europe based on molecular markers. J for Res 28:637–651. https://doi.org/10.1007/s11676-017-0393-8
Tóth EG, Bede-Fazekas Á, Vendramin GG, Bagnoli F, Höhn M (2019) Mid-Pleistocene and Holocene demographic fluctuation of Scots pine (Pinus sylvestris L.) in the Carpathian Mountains and the Pannonian Basin: Signs of historical expansions and contractions. Quatern Int 504:202–213. https://doi.org/10.1016/j.quaint.2017.11.024
Tulstrup N (1959) International trade in forest tree seed. Unasylva 13:196–201
Vasilyeva Y, Chertov N, Nechaeva Y, Sboeva Y, Pystogova N, Boronnikova S, Kalendar R (2021) Genetic structure, differentiation and originality of Pinus sylvestris L. populations in the east of the East European Plain. Forests 12:999. https://doi.org/10.3390/f12080999
Wachowiak W, Salmela MJ, Ennos RA, Iason G, Cavers S (2011) High genetic diversity at the extreme range edge: nucleotide variation at nuclear loci in Scots pine (Pinus sylvestris L.) in Scotland. Heredity 106:775–787. https://doi.org/10.1038/hdy.2010.118
Wachowiak W, Wόjkiewicz B, Cavers S, Lewandowski A (2014) High genetic similarity between Polish and North European Scots pine (Pinus sylvestris L.) populations at nuclear gene loci. Tree Genet Genomes 10:1015–1025. https://doi.org/10.1007/s11295-014-0739-8
Wachowiak W, Żukowska WB, Perry A, Lewandowski A, Cavers S, Łabiszak B (2022) Phylogeography of Scots pine in Europe and Asia based on mtDNA polymorphisms. J Syst Evol. https://doi.org/10.1111/jse.12907
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6:551–562. https://doi.org/10.1007/s10592-005-9009-5
Wójkiewicz B, Wachowiak W (2016) Substructuring of Scots pine in Europe based on polymorphism at chloroplast microsatellite loci. Flora – Morphology, Distribution, Functional Ecology of Plants 220: 142-149. https://doi.org/10.1016/j.flora.2016.03.005
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A 84:9054–9058. https://doi.org/10.1073/pnas.84.24.9054
Zimmer K, Sønstebø JH (2018) A preliminary study on the genetic structure of Northern European Pinus sylvestris L. by means of neutral nuclear microsatellite markers. Scand J for Res 33:6–13. https://doi.org/10.1080/02827581.2017.1337919
Żukowska WB, Wójkiewicz B, Litkowiec M, Wachowiak W (2017) Cross-amplification and multiplexing of cpSSRs and nSSRs in two closely related pine species (Pinus sylvestris L. and P. mugo Turra). Dendrobiology 77:59–64. https://doi.org/10.12657/denbio.077.005
Żukowska WB, Lewandowski A, Wójkiewicz B, Litkowiec M, Rożkowski R, Urbaniak L, Kowalczyk J (2023a) Changes in the gene pool composition of Scots pine depending on the mode of regeneration. Dendrobiology 89:46–55. https://doi.org/10.12657/denbio.089.005
Żukowska WB, Wójkiewicz B, Lewandowski A, László R, Wachowiak W (2023b) Genetic variation of Scots pine in Eurasia: impact of postglacial recolonisation and human-mediated gene transfer. Zenodo Repository V2:2023. https://doi.org/10.5281/zenodo.8405255. Accessed 04 Oct 2023
Acknowledgements
We thank members of the GreenTree consortium (EU Horizon 2020 Grant Agreement No. 676876) and NERC (Grant No. NE/H003959/1) for providing plant material from several populations included in the study. We also express our great gratitude to Dr Łukasz Walas from the Institute of Dendrology, Polish Academy of Sciences, for his help with demographic history modeling.
Code availability
Not applicable.
Consent to participate
Not applicable.
Funding
The research was financially supported by the Polish National Science Centre (UMO-2016/21/N/NZ9/01499) and the statutory activity of the Institute of Dendrology, Polish Academy of Sciences. WW acknowledges financial support from the Polish National Science Centre (UMO-2017/27/B/NZ9/00159).
Author information
Authors and Affiliations
Contributions
Conceptualization: WBŻ, WW; methodology: WBŻ, WW; formal analysis and investigation: WBŻ, BW; writing—original draft preparation: WBŻ; writing—review and editing: WBŻ, BW, AL, RL, WW; funding acquisition: WBŻ, WW; resources: AL, RL, WW. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that they obtained the approval of the Polish Ministry of Climate and Environment for conducting the study in protected areas in Poland.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling editor: Véronique Jorge.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix

Demographic scenarios tested with the ABC demographic history modeling. Pop1—Spain/France; Pop2 –Fennoscandia/Central Europe/Balkans; Pop3—Eastern Europe; Pop4—Urals/Asia

Mantel test results for all Scots pine populations analyzed in this study

Principal component analysis (PCA) carried out in DIYABC for the best demographic scenario (scenario 3)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Żukowska, W.B., Wójkiewicz, B., Lewandowski, A. et al. Genetic variation of Scots pine (Pinus sylvestris L.) in Eurasia: impact of postglacial recolonization and human-mediated gene transfer. Annals of Forest Science 80, 42 (2023). https://doi.org/10.1186/s13595-023-01207-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13595-023-01207-6