
Key message
Forest tree breeding must undergo significant revisions to adapt to the evolving challenges posed by climate change. Addressing the shifts in environmental conditions requires a comprehensive multidisciplinary approach that includes theoretical work and practical application. Specifically, there is a need to focus on developing new breeding strategies that are theoretically sound and practically feasible, considering the economic constraints of actual tree breeding programs. We present a novel concept utilizing genetic evaluation of multiple traits in forest stands of successive ages across wide ecological ranges. Incorporating genomics allows for detailed genetic evaluation, making use of high-density SNP markers and sophisticated algorithms like GBLUP for genetic parameter estimates. High-throughput phenotyping is conducted using drone-borne lidar technology to capture tree height and survival data across various forest stands. Assisted migration is considered to strategically position genotypes across predicted environmental climatic gradients, thereby accommodating the dynamic nature of ecological shifts. Mathematical optimization acts as an essential component for logistics, guiding the spatial allocation and timely substitution of genotypes to ensure a continually adaptive breeding program. The concept replaces distinct breeding cycles with continuous evaluation and selection, enhancing the rate of genetic response over time.
Similar content being viewed by others


1 Background
Genetic improvement of commercially important forest trees often involves long-term intensive recurrent selection programs requiring substantial resources and infrastructure. Breeding typically entails controlled pollinations creating structured pedigrees with offspring evaluated in replicated test sites within fixed ecological boundaries (zones). Production populations (typically seed orchards, SOs) are established at each selection cycle as sources of genetically superior forest reproductive material for afforestation (White et al. 2007).
Following the development of DNA markers, forest geneticists have proposed various ways to incorporate kinship analyses into their programs. The main incentive has been to avoid laborious control crosses and shorten generational intervals, thus boosting genetic response to selection. In addition, progeny testing can be simplified or entirely redirected to operational forest stands planted across broader environmental gradients. These advances alleviate the shortcomings of traditional programs that are spatially static and slow to respond to market demands and climate change (CC, Hanewinkel et al. 2013). Lambeth et al. (2001) suggested a technique called polymix breeding with parental analysis, which was designed to be more cost-effective than full-sib breeding and would enable the identification of male parents to control relatedness or create a full-sib mating pedigree. El-Kassaby and Lstibůrek (2009) extended this concept for open-pollinated populations, emphasizing that genetic gain could be made without making any crosses—an approach they termed Breeding without Breeding (BwB). Hansen and McKinney (2010) developed a quasi-field testing method to assign parentage of the Nordmann fir (Abies nordmanniana (Stev.) Spach.) offspring from a SO with 99 genotypes. Using 12 SSR markers and the parents’ genotype information, they could determine both parents for 98% of the progeny with 80% confidence.
Lstibůrek et al. (2017) proposed a “dynamic gene-resource landscape management combining utilization and conservation.” Based on the BwB methodology, the concept involves landscape-level genetic evaluation and selection of Norway spruce (Picea abies L.) from proven material exposed to realistic conditions over vast territories, i.e., across multiple environmental gradients. These advantages boost the genetic response to selection while conserving genetic resources. The first proof of the landscape-breeding concept was reported by Lstibůrek et al. (2020). They considered 4267 25- to 35-year-old European larch (Larix decidua Mill.) trees from 21 forest stands across four climatic regions in Austria. Individuals with high fitness and productivity attributes were identified using marker-based pedigree reconstruction and multi-trait, multi-site quantitative genetic analyses. As anticipated from an earlier comprehensive quantitative-genetic evaluation by Lstibůrek et al. (2015), the genetic parameters were found to be similar to those obtained using traditional “structured” pedigree methods. Poupon et al. (2021) employed response function methodology in the above larch in situ breeding program. They identified critical environmental gradients that impacted the studied traits and developed individual- and population-level response functions to estimate genetic variances along these gradients. Their concept provided an optimum allocation of the best-adapted reforestation material to the target location.
Lstibůrek et al. (2017, p. 2–3) offer an in-depth rationale for scaling up population sizes in breeding programs to address unpredictable CC and other risks. Critical attributes of “landscape” breeding programs encompass the preservation of rare alleles, augmentation of the breeder’s criterion through natural selection (facilitated by open pollination and the development of forest stands), detailed assessment of genotype × environment interactions across broad environmental gradients, etc.
2 Novel breeding concept
While landscape breeding methods are more advanced in tackling the challenges of CC, they still require ongoing revisions to maintain their effectiveness. The concept we introduce in this study is designed to address the main shortcomings and is built on the following key pillars.
-
1.
Given the fluctuating environmental conditions attributed to CC, it is essential to continually monitor the stability performance of individual genotypes across various forest stands and life stages. Utilizing semi-automated phenotyping and state-of-the-art genomic tools allows for the traits’ assessment throughout the life cycle of trees (Fig. 1). Unlike traditional methods, where progeny trials are usually established in a single year, this continuous evaluation exposes selection candidates to an ever-changing array of environmental conditions, both spatially and temporally. Multiple research projects are currently devoted to minimizing the cost of routine phenotyping and genotyping in forest stands, making the approach operationally feasible.
-
2.
Statistical-genetic evaluations should be conducted periodically, incorporating multiple traits, sites, and ages. This results in accurate estimates of genetic parameters and an updated ranking of selection candidates.
-
3.
Using mathematical optimization tools, a breeder can identify a set of the best-adapted individuals for potential inclusion in the SO (source of adapted forest reproductive material for afforestation).
-
4.
In the context of our proposed concept, the SO becomes a dynamic system where genetic improvement is continuous (a term “breeding arboretum” is introduced later for more precise terminology). Higher-ranking genotypes routinely replace inferior counterparts, optimizing the core subset of trees in their reproductive age (Fig. 2). Unlike the current breeding practices, the SO can, in theory, remain in a given location indefinitely. The genetic improvement of the SO crop is ongoing (continuous), with no distinct breeding cycles. Given the perpetual nature of this improvement, we anticipate a more rapid genetic response over time, which is not only crucial for adapting to CC but also economically beneficial.

Forest stands at a given site. This figure presents a simulated dataset for illustration. Each site consists of multiple forest stands established consecutively at 2-year intervals from a given parental source. Stands that have reached the selection age of 14 years are highlighted in red, indicating current selection candidates. The final harvest is planned for stands approaching an approximate height of 30 m. Statistical evaluation is performed across all life stages and stands, encompassing those yet to reach the selection age

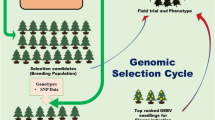
Rolling front landscape breeding. Phenotyping/genotyping in all stands (four assumed in the graphical illustration) is followed by combined multi-trait/site/age BLUP or GBLUP analysis, conducted periodically. Genetically superior individuals from the candidate population (exceeding the selection age) may enter the RESERVOIR subset of the arboretum. Genotypes in the RESERVOIR can either remain—with the possibility of either reaching reproductive age and joining the CORE subset or being removed during any evaluation stage. Genotypes in the CORE subset have two potential outcomes: they may either be retained or removed, with the latter occurring for reasons such as genetic inferiority or surpassing the reproductive age. Except for the initial period, the size of the CORE subset is dynamic, but it must meet a minimum \({N}_{CR}\). The quantity and specific identity of individuals shifted at each step (arrows) are periodically determined by mathematical optimization
The periodic evaluation and inclusion of the most adapted genotypes to the SO partly resemble the earlier Rolling Front approach (Borralho and Dutkowski 1998). Their idea was to proceed with controlled crosses between the best trees each year and establish new progeny trials with those from the prior year rather than waiting until all of that generation’s crosses were finished.
2.1 Analytical insights
The concept strategy takes into account multiple forest sites, each with unique environmental conditions suitable for genetic analysis and evaluation of respective genetic-by-environment interactions. These sites are spread across various environmental gradients and are established from a common first-generation clonal seed orchard (SO). At each of these sites, commercial forest stands are periodically established from bulk seed following natural mating (open-pollination) in the SO. The boundaries of these stands are recorded in the breeding organization’s database. In our model presentation (Fig. 1), a typical forest stand is established biennially at each site, although alternative time intervals could be considered. New seed lots from the SO will be utilized continuously, creating a time-based relation through a common set of parents and eventually, progenies. This approach gains significance as each annual seed crop offers a distinct genetic output, influenced by variables such as fertility fluctuations and pollen contamination (e.g., Funda and El-Kassaby 2012).
Individual offspring shall be monitored periodically in all assumed forest stands, utilizing a semi-automated protocol. Beyond traditional ground-level evaluations, the most feasible approach for site scanning is drone-borne LIDAR (Light Detection And Ranging). These sensors produce highly accurate, three-dimensional point clouds that can be easily processed into geopositioned rasters describing both the terrain and canopy height. Algorithms for automated single-tree detection are then employed to obtain individual tree heights and assess the survival of all candidates at every evaluation time point. Alternatively, cost-effective data collection can be achieved using RGB sensors, followed by 3D photogrammetry (Solvin et al. 2020). Further advancements in sensors, calibrations, and algorithms—for quantifying individual tree features like crown density or width, or health and adaptation traits from, for example, multispectral data—will enhance the efficiency of phenotyping.
DNA sample collection and subsequent genomic analyses may be tailored by the technological options in a given species and financial constraints. Random and top-phenotypic subsets are determined following the methodology in Lstibůrek et al. (2015). The random subset is used to estimate phenotypic and genetic variances to evaluate the top-phenotypic candidates afterward. Genetic evaluation options may range from a simple phenotypic evaluation (not requiring the random subset) to elaborate multi-trait/site/age REML-BLUP evaluation (identity-by-descent relationship matrix estimated by pedigree reconstruction as in Lstibůrek et al. 2020). Provided high-density SNP markers are available, the genomic (GBLUP) evaluation (Habier et al. 2007; VanRaden 2008) or its alternatives (Christensen et al. 2012; Legarra 2009) may be conducted with additional benefits, i.e., accounting for the Mendelian sampling term, capturing historical relationship patterns, and separating nonadditive genetic variances (e.g., El-Dien et al. 2016). These analyses consolidate all periodic measurements across multi-aged forest stands and multiple sites, enhancing the accuracy of genetic parameter estimates in subsequent evaluation stages. Although all stands across sites may be considered for the evaluation, only a subset has reached the selection age. Thus, during the periodic genetic re-evaluations, the candidate population expands to include additional superior individuals from stands that have reached this age threshold. In most forest tree species, inbreeding is detrimental, so complete avoidance or minimization of genetic relatedness in seed orchards (SOs) is typically advocated. In this framework, we assume all selected offspring in the initial cycle are unrelated, a claim that will be verified with kinship estimates during the evaluation process.
As previously discussed, the candidate population is periodically updated to include genetically superior individuals suited to current environmental conditions. Tree breeding relies on repeated cycles of genetic recombination, testing, and selection. Therefore, it is essential to place top-ranking individuals from the candidate population into a shared space where they can actively exchange gametes, i.e., reproduce. This could be again termed a seed orchard. However, we prefer the term “breeding arboretum” to emphasize that both breeding and deployment should occur in the same physical location. The arboretum is established by grafting the most superior genotypes from the candidate population. Each genotype is then represented by multiple clonal copies, known as ramets.
We propose dividing the breeding arboretum into two distinct compartments: the CORE and the RESERVOIR subsets, with NCR and NRS genotypes, respectively. Depending on species-specific logistics, these compartments can be spatially integrated or adjacent. The CORE subset is crucial for maintaining a requisite level of genetic diversity through reproductively active genotypes. Initially, all genotypes grafted during the first selection round remain in the arboretum until reaching reproductive age, in line with traditional SO establishment practices. However, since our strategy involves periodic reevaluation of forest stands and subsequent updates to the candidate population, the RESERVOIR subset serves a pivotal role. During each reevaluation, a mathematically optimized decision introduces new, genetically superior candidates from the updated population into the arboretum. Because grafts cannot be immediately replaced—they must first reach reproductive maturity—these new selections are initially placed in the RESERVOIR. Here, they remain until they mature enough to join the CORE subset. The RESERVOIR’s size is strategically designed to accommodate changes; it can both receive new candidates and remove them if they later prove to be genetically inferior. Fine-tuning the spatial arrangement between the CORE and RESERVOIR remains a topic for further investigation. For instance, expanding the spacing in the seed orchard may be necessary to mitigate competition among grafts of different ages. While a staggered clonal row system could simplify management (El-Kassaby et al. 2014), alternative strategies may better suit the particularities of a given species.
Eventually, all genotypes in the CORE subset will surpass their reproductive age and be progressively replaced by better candidates from the RESERVOIR. When operationally feasible, the arboretum can stay in the exact location, and the outlined process may continue as there are no distinct breeding cycles. In the later stages of the program, it will be beneficial to adopt the genomic relationship matrix to restrict the loss of genetic diversity and minimize inbreeding depression in the forest reproductive material originating from the arboretum (see Eq. 4 in section 3.1.2.). In addition, we advise considering the response function methodology (Poupon et al. 2021) to transfer genotypes across predicted climatic gradients. When adopting our concept across the landscape, multiple replications of the outlined system will eventually be established (or converted from existing SOs). It would then be possible to perform assisted migration along the gradients. Genetic coancestry will then be more easily controlled due to the periodic infusion of unrelated genotypes from distant sources.
As outlined above, the breeding arboretum serves as the primary source of seeds for establishing new stands, which are subsequently subject to genetic evaluation. This approach encompasses all elements of recurrent selection programs aimed at enhancing general combining ability—specifically, it focuses on increasing the frequency of favorable alleles through repeated cycles of genetic recombination, testing, and selection. It is worth noting that the forest stands chosen for evaluation represent only a small fraction of the commercial stands routinely established using seeds from the arboretum. In this sense, the arboretum fulfills its role as a conventional seed orchard. For clarity on essential terms related to the novel breeding concept, see Table 1.
3 Applicability and limitations
This manuscript presents a theoretical concept that has yet to be validated empirically. The intention was to initiate a scholarly discussion and lay the groundwork for future quantitative evaluations, potentially through computer simulations and species-specific pilot studies.
In the genomics context, our goal was to provide a range of options, from simple pedigree reconstruction using a limited set of SSR loci for coancestry control to more advanced GBLUP evaluations, as previously discussed. We opted not to include genomic prediction models because our strategy assumes that climatic conditions are in flux. Therefore, we rely on ongoing in situ monitoring of actual forest stands for evaluation. We do not claim that one approach is superior to the other; rather, these are distinct options that can be tailored to the specific needs of a given species. Factors for consideration include reproductive biology, deployment strategies, economic significance of a species, and rotation age, among others.
In all breeding programs, the phenotyping of a large number of individuals is constrained by limited budgets. This also narrows the range of genetic entries that can be tested, ultimately forcing a trade-off among selection intensity, multi-trait selection, and genetic diversity. We suggest a streamlined approach for phenotyping, as the main goal is to improve performance over time by leveraging individual variation in response to climatic fluctuations. Drone-borne sensors are most effective in scenarios featuring even-aged monocultures that are fairly evenly spaced. Consequently, stand management remains crucial, similar to conventional progeny testing. Automatic species detection in mixed-species stands is still too inaccurate, making these situations complex to manage. Uneven-aged stands present further challenges, as current sensor technology lacks the capability to determine tree age.
Seed orchard managers may be reluctant to continuously swap clones in the orchard, as this creates an uneven situation where young grafts are overshadowed by older ones. For many species, these younger grafts have a reduced gametic contribution, which must be factored into estimates of the effective population size. Moreover, the orchard’s production capacity may temporarily decline when older, more productive grafts are replaced. However, this situation is likely to reverse as the fertility of the oldest grafts declines, making them candidates for replacement—commonly occurring after 40-60 years in species like Norway spruce and Scots pine (Almqvist et al. 2010).
3.1 Mathematical optimization
We propose a linear mixed-integer mathematical programming model that offers room for further expansion (one could draw on insights from sources such as Funda et al. 2009). The model facilitates decision-making according to the description above and Fig. 2.
3.1.1 Objective function
The optimization aims to maximize genetic response to selection.
where \({a}_{i}\) is the breeding value of the ith genotype. Binary variable \({x}_{i}\) (the optimization output) designates whether a given genotype should be included in the breeding arboretum (value 1) or otherwise (value 0). \({N}_{CN}\) is the number of genotypes in the candidate population. Substitution of an existing genotype in the arboretum by a superior candidate genotype is associated with a cost. Breeders may calculate a constant weight using the cost-benefit analysis. The weight can be easily added to the breeding values of genotypes that are currently present in the arboretum.
3.1.2 Constraints
The following constraint will ensure that the number of selected genotypes will meet the size of the breeding arboretum.
where \({N}_{CR}\) and \({N}_{RS}\) and the respective number of genotypes in the CORE and RESERVOIR subsets.
The following constraint will ensure that the CORE subset consists of a minimum number of genotypes within the reproductive age.
where \({t}_{i}\) is an input binary constant designating whether the ith genotype is within its reproductive age (value 1) or otherwise (value 0).
The following set of constraints ensures that all selections are unrelated (maximum parental contribution equal to 1). For example, let’s assume parent 1.
where \({y}_{1i}\) is a binary input constant designating whether individual 1 is the respective parent of the offspring i. The above constraint must be satisfied across all parents, i.e., one equation per parent. Provided that the genomic relationship matrix should be used to constrain selection, one may adopt one of the algorithms reviewed by Woolliams et al. (2015).
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Almqvist C, Wennström U. Karlsson B (2010) Förädlat skogsodlingsmaterial 2010-2050 (Redogörelse från Skogforsk). Skogforsk
Borralho NM, Dutkowski GW (1998) Comparison of rolling front and discrete generation breeding strategies for trees. Can J For Res 28:987–993
Christensen OF, Madsen P, Nielsen B, Ostersen T, Su G (2012) Single-step methods for genomic evaluation in pigs. Animal 6:1565–1571. https://doi.org/10.1017/S1751731112000742
El-Dien OG, Ratcliffe B, Klápště J, Porth I, Chen C, El-Kassaby YA (2016) Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3: Genes Genomes, Genetics 6:743–753. https://doi.org/10.1534/g3.115.025957
El-Kassaby YA, Lstibůrek M (2009) Breeding without breeding. Genet Res 91:111–120. https://doi.org/10.1017/S001667230900007X
El-Kassaby YA, Fayed M, Klápště J, Lstibůrek M (2014) Randomized, replicated, staggered clonal-row (R2SCR) seed orchard design. Tree Genet Genomes 10:555–563. https://doi.org/10.1007/s11295-014-0703-7
Funda T, El-Kassaby YA (2012) Seed orchard genetics. CABI Rev 7:13. https://doi.org/10.1079/PAVSNNR20127013
Funda T, Lstibůrek M, Lachout P, Klápště J, El-Kassaby YA (2009) Optimization of combined genetic gain and diversity for collection and deployment of seed orchard crops. Tree Genet Genomes 5:583–593. https://doi.org/10.1007/s11295-009-0211-3
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397
Hanewinkel M, Cullmann DA, Schelhaas MJ, Nabuurs GJ, Zimmermann NE (2013) Climate change may cause severe loss in the economic value of European forest land. Nat Clim Change 3:203–207. https://doi.org/10.1038/nclimate1687
Hansen OK, McKinney LV (2010) Establishment of a quasi-field trial in Abies nordmanniana—test of a new approach to forest tree breeding. Tree Genet Genomes 6:345–355. https://doi.org/10.1007/s11295-009-0253-6
Lambeth C, Lee BC, O’Malley D, Wheeler N (2001) Polymix breeding with parental analysis of progeny: an alternative to full-sib breeding and testing. Theor Appl Genet 103:930–943
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663. https://doi.org/10.3168/jds.2009-2061
Lstibůrek M, Hodge GR, Lachout P (2015) Uncovering genetic information from commercial forest plantations—making up for lost time using “Breeding without Breeding.” Tree Genet Genomes 11:55. https://doi.org/10.1007/s11295-015-0881-y
Lstibůrek M, El-Kassaby YA, Skrøppa T, Hodge GR, Sønstebø JH, Steffenrem A (2017) Dynamic gene-resource landscape management of Norway spruce: combining utilization and conservation. Front Plant Sci 8:1810. https://doi.org/10.3389/fpls.2017.01810
Lstibůrek M, Schueler S, El-Kassaby YA, Hodge GR, Stejskal J, Korecký J, Škorpík P, Konrad H, Geburek T (2020) In situ genetic evaluation of European larch across climatic regions using marker-based pedigree reconstruction. Front Genet 11:28. https://doi.org/10.3389/fgene.2020.00028
Poupon V, Chakraborty D, Stejskal J, Konrad H, Schueler S, Lstibůrek M (2021) Accelerating adaptation of forest trees to climate change using individual tree response functions. Front Plant Sci 12:758221. https://doi.org/10.3389/fpls.2021.758221
Solvin TM, Puliti S, Steffenrem A (2020) Use of UAV photogrammetric data in forest genetic trials: measuring tree height, growth, and phenology in Norway spruce (Picea abies L. Karst.). Scand J For Res 35:322–333. https://doi.org/10.1080/02827581.2020.1806350
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
White TL, Adams WT, Neale DB (2007) Forest genetics. CAB International, Oxfordshire, UK
Woolliams JA, Berg P, Dagnachew BS, THE Meuwissen (2015) Genetic contributions and their optimization. J Anim Breed Genet 132:89–99. https://doi.org/10.1111/jbg.12148
Acknowledgements
Not applicable.
Funding
The research leading to these results has received funding from the EEA/Norway Grants 2014–2021 and the Technology Agency of the Czech Republic.
Author information
Authors and Affiliations
Contributions
The authors contributed to the text and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors gave their informed consent to this publication and its content.
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Marjana Westergren.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lstibůrek, M., García‐Gil, M.R. & Steffenrem, A. Rolling front landscape breeding. Annals of Forest Science 80, 36 (2023). https://doi.org/10.1186/s13595-023-01203-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13595-023-01203-w