
Abstract
Streptococcus ruminantium is the causative agent of several bovine and ovine diseases, however reports are uncommon and application of whole genome sequencing to identify is rare. We report for the first time, a severe ovine mastitis outbreak caused by S. ruminantium in Italy, 2022. S. ruminantium was isolated from 12 adult lactating ewes with diffuse nodules in the mammary parenchyma and predominantly serous and clotted milk. All outbreak isolates, along with five additional historical Italian isolates (between 2011 and 2017), were genomically characterised and then analysed in the context of all publicly available S. ruminantium genomes. Antimicrobial susceptibility testing was performed to determine the MICs of 16 antibiotics. The results showed that all isolates were susceptible to all antimicrobials tested except kanamycin. Single Nucleotide Variant analysis confirmed this as a clonal outbreak across 10 sheep (≤ 15 SNVs), while the two others were colonised by more distantly related clones (≤ 53 pairwise SNVs), indicating the presence of multiple infecting lineages. The five historical S. ruminantium isolates were comprised of genetically-distant singletons (between 1259 and 5430 pairwise SNVs to 2022 outbreak isolates). Ovine isolates were found to be genetically distinct to bovine isolates, forming monophyletic groups. Bovine isolates were similarly made up of singleton clones in all but two isolates. Taken together, our genomic analysis using all globally available genomes is consistent with general opportunistic pathogenesis of S. ruminantium. We encourage future genomic surveillance efforts to facilitate outbreak detection, as well as improve our understanding of this poorly-understood, multi-host, zoonotic pathogen.
Similar content being viewed by others

Introduction
Mastitis is one of the most common and costly diseases of dairy sheep and goats. Infectious mastitis outbreaks can be caused by a variety of bacterial species, including the genera Staphylococcus and Streptococcus [1,2,3]. In both genera, new species are constantly being discovered or renamed through the application of genomic analysis. Streptococcus (S.) ruminantium was first proposed as a name for a novel species in 2017 by Tohya et al. [4], based on phylogenetic analysis of 16S rRNA and sodA gene sequences. S. ruminantium was previously classified as serotype 33 of S. suis, one of the most important swine pathogens responsible for various diseases including pneumonia, arthritis, meningitis and endocarditis [5]. It is also a zoonotic agent that affects humans in close contact with infected pigs or pig products [6]. As it is difficult to differentiate S. suis from S. ruminantium in routine diagnostics based on the use of biochemical tests, either whole genome sequencing or specific PCRs can be used for differential identification [7,8,9,10,11].
Following the classification of S. ruminantium as a new species, five case reports associated with this species in ruminants have been published along with 24 publicly available genomes. Okura et al. [11] reported the isolation of S. ruminantium from cattle with endocarditis, arthritis and pneumonia or respiratory disease in Japan, while Gottschalk et al. [12] described the bacterial isolation from 10 cattle and 4 sheep with arthritis, pneumonia, endocarditis and abscesses in Canada. Both reports lacked information on clinical signs and in some cases have not made sequence data publicly available. Recently, S. ruminantium was associated with pathology in 4 Pyrenean chamois and 1 domestic sheep from NE Spain [13]. In wild ruminants, S. ruminantium has been isolated from lung lesions and heart valve vegetations, and in domestic ruminants from liver abscesses [13].
The present study provides the first information on the association of S. ruminantium with an outbreak of mastitis in dairy ewes in Sardinia, Italy. We describe the clinical signs of the animals, the isolation and identification of the bacterial species associated with mastitis. All S. ruminantium isolates were subjected to molecular and genome sequence analysis to gain insight into bacterial clonality, pathogenicity and the presence of antimicrobial resistance genes.
Materials and methods
Farm and clinical examination
The owner of the dairy sheep farm, located in the eastern part of Sardinia, contacted the Istituto Zooprofilattico Sperimentale della Sardegna (IZSSA) in 2022 due to several cases of clinical mastitis. Following contacts with the farmer and the farm veterinarian, an outbreak investigation was initiated to detect and identify the microorganism, a possible source of the organism and to control the outbreak.
During the farm visit (31/05/2022), information was obtained from the farmer on farm type, flock size, milking and hygiene practices including use of disinfectants and antibiotics, previous vaccinations, and type of milking (manual or mechanical). Clinical examination was performed on each half-udder and included a general inspection, assessment of half-udder consistency, macroscopic examination of milk and palpation of supramammary lymph nodes. During the examination, the presence of pustules, crusts, corneal growths, ulcers, nodules, abscesses, rubor, calor and dolor (by warmth on palpation) was assessed. The consistency of the udder on palpation was classified as normal, edematous, sclerotic, or atrophic. The macroscopic appearance of the milk was classified as: normal, serous, haemorrhagic, presence of clots or absence of secretion [14].
Out of the 174 lactating ewes, 24 were found to have clinical mastitis. Twelve ewes were selected based on common udder clinical pictures, and milk and blood samples were taken from these ewes.
Milk sampling and microbiological cultures
From each of the 12 selected ewes, we collected at least 5 ml of milk from both half-udders in a sterile container, after cleaning the teat with 90% denatured ethanol and discarding the first drops of milk in a bucket containing sodium hypochlorite. Samples were refrigerated and subjected to microbiological examination in the laboratory within 12 h from collection. For microbiological cultures, 10 μL of milk were seeded in 5% sheep blood agar and incubated at 37 ± 1 °C for 24–48 h. Only pure bacterial colonies grew in the 12 blood agar plates. All isolates were identified at the genus level based on growth characteristics, morphology of colonies and microscopic examination after Gram staining.
MALDI-TOF MS identification
In this study, we analysed all 12 outbreak isolates, two S. suis isolates (3089 and 3627) collected from pigs with pneumonia, and five historical isolates (OM1195, OM2067, OM2267, OM2774 and OM3442) belonging to our bank and collected from sheep mastitis in previous years (Additional file 2). These historical isolates, archived as S. suis, were re-identified to understand whether the circulation of S. ruminantium in Sardinia was before 2022. For MALDI-TOF MS analysis, the direct colony transfer protocol was applied. Each target plate included two spots of Bacterial Test Standard (Bruker Daltonik GmbH). Mass spectra were acquired using the MALDI-TOF MS spectrometer in a linear positive mode (MALDI Biotyper Sirius One, Bruker Daltonics). The MBT Compass library revision K (2022), covering 4274 species/entries, were used for bacterial identification (Additional file 1). The similarity of patterns was represented as a score, according to manufacturer specifications: a score value of < 1.7 indicated that identification was not reliable; scores between 1.7–2.0 that identification was reliable at the genus level; scores between 2.0 and 2.3 that identification was reliable at the genus level and probable at the species level; scores higher than 2.3 indicated highly probable species identification.
DNA extraction, polymerase chain reaction (PCR) amplification, and restriction fragment length polymorphism (RFLP) analysis
Genomic DNA was extracted from the 19 (12 outbreak, 2 S. suis and 5 historical) isolates as described by Onni et al. [15], while species identification was based on PCR amplification of the glyceraldehyde-3-phosphate dehydrogenase gene (gap) gene (Table 1). Fifteen microliters of both PCR amplifications were digested in a 30 µL volume containing 10 × FastDigest Green buffer, 0.25 µL of 20 mg/mL acetylated BSA, and 1 µL of FastDigest AluI enzyme (Thermo Scientific, CA, USA). Reaction mixtures were incubated at 37 °C for 15 min and directly loaded on the precast NuPAGE™ gels (Invitrogen).
Amplicon sequencing, PCR for S. ruminantium and pulsed field gel electrophoresis (PFGE)
The gap gene amplicons of all 19 isolates were sequenced at BMR Genomics with the Sanger sequencing option. The nucleotide sequences were compared to sequences in the GenBank database using the Basic Local Alignment Search Tool (BLAST).
The primers used for S. ruminantium identification are listed in Table 1. After PCR reactions, amplicons were analyzed by electrophoresis on agarose gels.
Isolates were genotyped by PFGE. DNA was extracted with Bio-Rad CHEF genomic DNA plug kit (BioRad, Segrate, Italy) according to the manufacturer’s instructions. Each plug was digested with 20 U of SmaI (Roche) for 3 h at 25 °C. Electrophoresis was carried out in a contour-clamped homogeneous electric field (CHEF)- Mapper system (BioRad) at 14 °C in 0.5 × TBE buffer. DNA fragments were separated after 18 h migration with 6 V/cm, 120° at pulse times of 10–45 s. The digitalized PFGE patterns were analyzed with the Gel Compar II software (Applied Maths, Sint-Martens-Latem, Belgium).
Antimicrobial susceptibility testing
The Minimum Inhibitory Concentration (MIC) of 16 antimicrobial agents were determined by the broth microdilution method using the Sensititre™ ITISVE8 plates (Thermo Fisher Scientific, West Sussex, UK). The customized plates were prepared for Istituto Zooprofilattico della Lombardia e Emilia Romagna to test the antimicrobial susceptibility of Gram-positive bacteria responsible of ovine and caprine mastitis. The antimicrobials tested were: amoxicillin/clavulanic acid 2:1 ratio (AUG2, 16/8–0.25/0.12 µg/mL), enrofloxacin (ENRO, 4–0.25 µg/mL), cefazolin (FAZ, 8–0.25 µg/mL), ceftiofur (XNL, 8–0.25 µg/mL), erythromycin (ERY, 8–0.03 µg/mL), florfenicol (FFN, 8–2 µg/mL), tetracycline (TET, 16–0.25 µg/mL), kanamycin high level (KAN HL, 500–250 µg/mL), kanamycin (KAN, 32–8 µg/mL), trimethoprim/sulfadiazine (TBR, 8/152–0.12/2.38 µg/mL), oxacillin + 2% NaCl (OXA + , 4–0.25 µg/mL), rifampin (RIF, 2–0.06 µg/mL), clindamycin (CLI, 2–0.5 µg/mL), sulfisoxazole (FIS, 512–128 µg/mL), ampicillin (AMP, 16–0.03 µg/mL), tilmicosin (TIL, 32–8 µg/mL), and penicillin (PEN, 16–0.03 µg/mL). The MIC test conditions were performed according to the manufacturer’s instructions. Plates were incubated at 37 °C for 24 h and the reading was performed manually. Where possible, results were interpreted according to CLSI breakpoints from other animals or other Streptococcus species [17], otherwise the EUCAST range was used [18]. All 17 (12 outbreak and 5 historical) S. ruminantium isolates used in this study, antimicrobials tested and their breakpoints are listed in Additional file 2.
Whole-genome sequencing
Genomes from all 17 S. ruminantium isolates were prepared using the enzymatic lysis method as described previously [19]. Sequencing libraries were made using Ion Xpress™ Plus Fragment Library Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Libraries were sequenced with an IonTorrent Personal Genome Machine (PMG) (Life Technologies, Carlsbad, CA) at the IZSSA.
Genome acquisition and assembly
For data generated in this study, SPAdes [20] version 3.15.3 was used to assemble the IonTorrent reads with the following options: “–iontorrent –isolate”. K-mers were iterated through starting with −k 27, then 53,71,87,99,111,119,127 with the “–restart-from k[53…127]”. After these 8 assemblies were complete, the.gfa files were evaluated using GFA-dead-end-counter version 1.0.0 [21]. Assemblies with the fewest Graphical Fragment Assembly (GFA) dead ends, then fewest contigs were chosen as the best assembly. Assemblies with > 20 GFA dead ends were re-sequenced.
For data obtained from other studies, sequencing reads were obtained from Sequence Read Archive where possible, or as assemblies from Genbank if raw reads were unavailable. In total, 3 readsets and 21 assemblies were obtained from three studies [11, 22, 23]. For genomes with long and short reads available, long reads were assembled using Flye version 2.8.1-b1676 [24] with the following options: “–nano-raw -g 2100000–asm-coverage 80 –plasmids”. Assemblies were long read polished using medaka version 1.8.0 with the following option: “medaka_consensus −m r941_min_high_g360”. Paired short reads were then trimmed with Trim Galore version 0.5.0 using the −q 20 option [25]. BWA-MEM version 0.7.17 [26] was used to align each paired short read file to the long read assembled genome, then Polypolish version 0.5.0 was used for short read polishing [27].
Average nucleotide identity
To confirm the species identification, Average Nucleotide Identity (ANI) was calculated in a pairwise fashion between all isolates identified in this study and all known S. ruminantium genomes, along with Streptococcus suis (accession: GCF_000026745.1) using FastANI version 1.33 [28] with “–fragLen 1000”.
Single nucleotide variant analysis
Single Nucleotide Variant (SNV) analysis was used to determine outbreak potential of isolates. This was performed using SKA version 1.0.0 [29] using the “ska fasta”, “ska distance -s 20” then “ska compare” options. SNV thresholds were determined by plotting a histogram of the pairwise SNVs between potentially linked isolates.
Isolate clustering
PopPUNK version 2.4.0 [30] was used to assign genomes to clusters. The “create-db” function was used with the following options: “–sketch-size 1 000 000 –min-k 15 –max-k 29 –qc-filter prune”. Then the “fit-model” function was used with the following options: “bgmm -K 5 –ranks 1,2,3,5 –graph-weights”. Then the “fit-model” function was used with the following options: “refine –graph-weights –unconstrained”. Then the “poppunk_visualise” function was used, with the “—distances” and “–previous-clustering” utilising the refined model fit, to output a neighbour-joining core tree.
Genome annotation
Bakta version 1.5.1 [31] was used to annotate all remaining genomes. Bakta database accessed on 18/08/2022).
Antibiotic resistance screening
AMRFinderPlus version 3.10.23 [32] was used to screen assemblies for antibiotic resistance genes using protein input with identity set to 80%: “-i 0.8”.
Pangenomics
The pangenome was constructed using Panaroo version 1.2.8 [33] with the following options: “–clean-mode sensitive -a core –aligner mafft –no_clean_edges –core_threshold 0.98 –merge_paralogs –remove-invalid-genes”.
Putative virulence factor screen
To identify putative virulence factors within S. ruminantium genomes, a custom database was constructed using the abricate command “—setupdb”. Sequences were obtained using closely-related Streptococcus suis studies [34, 35], then duplicates removed. This is available as Additional file 3. Blastp (Blast + version 2.9.0 [36]) was used with the following options: blastp -outfmt “6 qseqid sseqid pident length qcovs” “-max_hsps 1 -num_alignments 1” then filtered on 80% query coverage and identity to identity positive hits.
Visualisation
Visualisation was performed in R version 4.1.2 [37], RStudio version 1.4.1717 [38], with the following software packages: tidyverse version 1.3.1 [39], RColorBrewer version 1.1–2 [40], igraph version 1.2.7 [41], ggraph version 2.0.5 [42], aplot version 0.1.6 [43], ape version 5.5 [44], ggtree version 3.7.1.003 [45], cluster version 2.1.2 [46] ggforce version 0.3.3 [42]. All R code can be found in Additional file 4.
Data availability
All assemblies and reads have been uploaded to Genbank under BioProject Accession PRJNA1009676. BioSample accessions can be found in Additional file 2.
Results
Flock and clinical examination
The Sardinian flock, consisting of 174 adult lactating ewes, was of the semi-wild type with the co-presence of other animal species such as horses and pigs but no cows. The ewes were grouped into a clean environment for milking only. The trolley milking machine was equipped with a single bucket, two teat cups, and a vacuum pump. The parameters set were: vacuum (−41 kPa), pulsation (180) and percentage between vacuum and atmospheric pressure at the teat (50%). The main palpable clinical signs in the 12 sheep with mastitis are shown in Table 2. The most common findings were enlarged supramammary lymph nodes and the presence of indicators of chronic infection such as nodules, sclerosis and atrophy of half or both mammary parenchyma. However, an overlap between chronic and acute infection was found in almost all animals, due to the simultaneous presence of clinical signs such as rubor, calor, dolor, edema and the presence of blood in the milk. Nine out of the 16 milk samples were serous with clots. No antimicrobial treatment was given to infected sheep.
Molecular identification of S. ruminantium
Prior to whole genome sequencing, an attempt was made to identify the 12 outbreak isolates by MALDI-TOF MS, but the result was “No Organism Identification Possible”, with a score < 1.33. RFLP also showed distinct profiles of these 12 isolates compared to two field isolates (3089 and 3627) collected from pig lung lesions and identified as S. suis by MALDI-TOF MS with a score > 2.3 (Additional file 5). PCR amplification and sequencing of the gap gene showed 97% identity and 99% query coverage to S. ruminantium (accession: CP019557.1) (Additional file 6).
All the 12 S. ruminantium isolates and the two S. suis isolates were PCR positive for the gdh gene, whereas all S. ruminantium isolates were negative for S. suis-specific recN-PCR, while positive for S. ruminantium-specific 16S rRNA-PCR (Additional file 7). The historical isolates showed the same characteristics (MALDI-TOF, RFLP and PCR) as the 12 outbreak S. ruminantium isolates.
Molecular identification of the 17 S. ruminantium was confirmed by whole-genome sequencing and average nucleotide identity (ANI), with all isolates falling within 98.3% ANI of 21 published S. ruminantium genomes.
Outbreak analysis
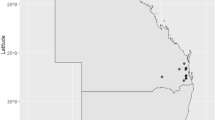
Initially, PFGE was performed to rapidly assess the degree of genetic diversity of the 12 S. ruminantium isolates, which indicated a high degree of genetic similarity and thus spread within the flock (Additional file 8). Pairwise SNV analysis after whole genome sequencing confirmed this finding at higher resolution. Comparison of all pairwise SNVs between the 12 Sardinian isolates suspected to be part of an outbreak showed three clear SNV boundaries, first at ≤ 13 SNVs, then between ≥ 30 to ≤ 53 SNVs, then > 53 SNVs (Additional files 9 and 10). Given the epidemiological context of this event, that all isolates were collected on a single day from the same Sardinian flock, and the distribution of pairwise SNVs, direct transmission was set at ≤ 15 SNVs. The SNV network provides strong evidence that a clonal outbreak cluster of 10 sheep occurred on this farm in 2022 (Figure 1). The presence of two more distantly related isolates (within 53 pairwise SNVs to the other 2022 isolates) indicated the presence of multiple infecting lineages or lineages that had recently diverged. This suggests that different S. ruminantium lineages may be more prevalent in sheep populations than previously thought. Historical isolates collected throughout Sardinia between 2011 and 2017 were genetically distant from the 2022 outbreak isolates (1259–5430 pairwise SNVs) (Additional file 10).

Pairwise single nucleotide variant (SNV) network. Isolates are shown as nodes, coloured by year of isolation. Transmission was set at ≤ 15 SNVs (solid lines), with more distantly related isolates found with ≤ 53 SNVs (dotted line). Isolates from previous years with ≥ 1000 pairwise SNVs have blank lines.
Antimicrobial susceptibility and resistance gene determinants
All the 17 S. ruminantium isolates were susceptible to AUG2, FAZ, XNL, FFN, TBR, OXA + , RIF, CLI, FIS, AMP, TIL and PEN, as no growth was observed at the lowest concentration of antimicrobial tested. MIC values were determined for TET and ENRO, which can be considered as susceptible breakpoints according to the guidelines described in Materials and methods. Isolates grew at all the lowest concentrations of KAN tested (32-16-8 µg/mL) but not at the highest (500–250 µg/mL). (Additional file 2). Based on the AMRFinderPlus analysis (dated 31/07/2023), no antimicrobial resistance genes were found in the S. ruminantium isolates (Additional file 11).
Cow and sheep isolates are genomically distinct
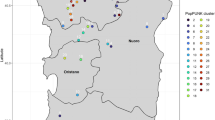
The lineages of these isolates were then analysed in the context of all publicly available S. ruminantium genomes (Figure 2). The Sardinian sheep isolates isolated during this study clustered separately from the Japanese cow isolates and had a SNV mean of 7934 (range 7521–8226), indicating considerable divergence. Due the limited number of genomes currently available, it is unclear if this is a regional or host-specific observation. Notably, Japanese cow isolates were made up of distinct singleton lineages, except for two isolates (S. ruminantium DTK284 and DTK285) which were part of the same cluster (6 SNVs apart) despite being isolated 6 years apart. The phylogeny was also concordant with the SNV outbreak analysis regarding the 12 Sardinian isolates, where the SNV-linked outbreak (10 isolates) was clustered together with short branches, while also being part of the same PopPUNK cluster.

Neighbour-joining tree of all publicly available S. ruminantium genomes. Isolates coloured by PopPUNK cluster and source shown by tip shape.
To determine any host-specific gene content, the pangenome of all S. ruminantium genomes was then analysed. A core genome of 1571 from of a total 4178 pan genes was identified (95% threshold), indicating a highly conserved gene set of 37.6% across all isolates. Forty-one genes were found to be specifically conserved in sheep isolates (Additional file 12). These mostly consisted of mobile genetic elements including transposes (4), integrases (2), phage proteins (7), as well as transcriptional regulator/DNA binding (8) and several metabolism and secondary metabolite-related genes (8). Three genes were found to be specifically conserved in cow-only isolates, consisting of mdlB, a ABC multidrug transport system and two hypothetical proteins.
Virulence genes
Putative virulence genes were screened using those identified in closely-related species Streptococcus suis. Most were core across all isolates (Figure 3 and Additional file 13) regardless of source of isolation, however there were a few notable exceptions. A 3-ketoacyl-ACP reductase (WP_032511883.1), laminin binding protein (WP_012774966.1) and glutamate dehydrogenase (WP_011921833.1) were found to be almost always present within cow isolates but absent in sheep mastitis isolates. Cow isolates were missing citB (CAZ51628.1) cps9E (AAF18948.1) and bgaC, a surface-anchored beta-galactosidase (WP_011922008.1).

Heatmap showing presence and absence of putative virulence factors in all S. ruminantium genomes used in this study. Both axes were hierarchically clustered via Gower dissimilarity.
Discussion
These data provide the first global genomic insights into this multi-host ruminant pathogen. The presence of discrete, singleton lineages found to be the cause of pathogenesis in cows is consistent with the historical Sardinian sheep isolates in that these are likely to be opportunistic pathogens, as noted in other studies of S. ruminantium [11]. However, our study is the first to link S. ruminantium to a demonstrable outbreak. The presence of historical S. ruminantium isolates, initially identified as S. suis and collected between 2011–2017, indicates that this pathogenic species has been present in the region for over a decade. The clonal linkage of Japanese cow isolates S. ruminantium DTK284 and DTK285, isolated 6 years apart, shows that different clones can remain in circulation, but overwhelmingly these appear to be opportunistic infections caused by genetically distinct clones in both cattle and sheep. While mastitis is a serious clinical condition, there have been no S. ruminantium-associated endocarditis, respiratory disease, or death in sheep (Table 2), as has been reported in cows [4, 11, 22]. However, although comparisons were made between sheep and cow isolates in this study, they should be treated with caution due to the small number of genomes available. Given the severity of the disease in ruminants and the ability to spread clonally within a herd, surveillance efforts should be increased to monitor this important agricultural pathogen.
All Sardinian sheep isolates were resistant to the aminoglycoside tested, kanamycin, while being susceptible to all other classes tested (Additional file 2). This is broadly in agreement with a previous study of S. uberis mastitis in Sardinian sheep, which also showed high percentage of intrinsic resistance to aminoglycosides, but sensitivity to other antibiotics [19]. Notably, the historical isolates from 2016 onwards showed a similar antibiotic resistance profile to that of the 2022 outbreak. Characterization of these genetic resistance determinants is critical for future and informed surveillance of S. ruminantium and closely related pathogens. In contrast, many resistance genes were found in 16/24 Japanese cow isolates (median number of subclass resistances = 3). tet(M) or tet(O) tetracycline resistance was the most common (15 isolates), followed by ant(6)-Ia or aadE streptomycin resistance (12 isolates) then erm(B) macrolide resistance (10 isolates) (Additional file 11). The significant levels of antibiotic resistance, likely reflecting the use of these or related antimicrobials in that region [47].
At least 64 putative virulence genes were identified in S. suis that were present in each S. ruminantium genome in this study, further demonstrating their relatively recent divergence [4] and pathogenic overlap. Given that S. suis can also cause mastitis [48] and meningitis in cattle [49] and has previously been isolated by the Istituto Zooprofilattico Sperimentale della Sardegna during routine surveillance of sheep mastitis (unpublished data), it is likely that these pathogens share a similar opportunistic pathogenic strategy.
Future genomic sequencing of clinical isolates is recommended so that a greater number of genomes can be used to improve future genomic understanding of this under-represented but relevant agricultural pathogen. Short-term but frequent gastrointestinal colonisation and faecal shedding by the closely related S. uberis has been demonstrated [50] and it is possible that S. ruminantium follows a similar strategy. Therefore, we recommend molecular and/or genomic surveillance of non-clinical (healthy) host gastrointestinal tracts to determine non-clinical carriage rates, which would help to determine flock/herd susceptibility and allow early intervention to prevent disease onset, while flock outbreaks could be prevented by isolation of positive hosts.
References
Marogna G, Rolesu S, Lollai S, Tola S, Leori SG (2010) Clinical findings in sheep farms affected by recurrent bacterial mastitis. Small Rumin Res 88:119–125. https://doi.org/10.1016/j.smallrumres.2009.12.019
Gelasakis AI, Mavrogianni VS, Petridis IG, Vasileiou NGC, Fthenakis GC (2015) Mastitis in sheep: the last 10 years and the future of research. Vet Microbiol 181:136–146. https://doi.org/10.1016/j.vetmic.2015.07.009
Dore S, Liciardi M, Amatiste S, Bergagna S, Bolzoni G, Caligiuri V, Cerrone A, Farina G, Montagna CO, Saletti MA, Scatassa ML, Sotgiu G, Cannas EA (2016) Survey on small ruminant bacterial mastitis in Italy, 2013–2014. Small Rumin Res 141:91–93. https://doi.org/10.1016/j.smallrumres.2016.07.010
Tohya M, Arai S, Tomida J, Watanabe T, Kawamura Y, Katsumi M, Ushimizu M, Ishida-Kuroki K, Yoshizumi M, Uzawa Y, Iguchi S, Yoshida A, Kikuchi K, Sekizaki T (2017) Defining the taxonomic status of Streptococcus suis serotype 33: the proposal for Streptococcus ruminantium sp. nov. Int J Syst Evol Microbiol 67:3660–3665. https://doi.org/10.1099/ijsem.0.002204
Gottschalk M, Segura M (2019) Streptococci. In: Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW, Zhang J (eds) Diseases of swine. John Wiley & Sons, Hoboken
Lun ZR, Wang QP, Chen XG, Li AX, Zhu XQ (2007) Streptococcus suis: an emerging zoonotic pathogen. Lancet Infect Dis 7:201–209. https://doi.org/10.1016/S1473-3099(07)70001-4
Ishida S, Tien LHT, Osawa R, Ma T, Nomoto R, Kawamura Y, Takahashi T, Kikuchi N, Kikuchi K, Sekizaki T (2014) Development of an appropriate PCR system for the reclassification of Streptococcus suis. J Microbiol Methods 107:66–70. https://doi.org/10.1016/j.mimet.2014.09.003
Nomoto R, Maruyama F, Ishida S, Tohya M, Sekizaki T, Osawa R (2015) Reappraisal of the taxonomy of Streptococcus suis serotypes 20, 22 and 26: Streptococcus parasuis sp. nov. Int J Syst Evol Microbiol 65:438–443. https://doi.org/10.1099/ijs.0.067116-0
Hill JE, Gottschalk M, Brousseau R, Harel J, Hemmingsen SM, Goh SH (2005) Biochemical analysis, cpn60 and 16S rDNA sequence data indicate that Streptococcus suis serotypes 32 and 34, isolated from pigs, are Streptococcus orisratti. Vet Microbiol 107:63–66. https://doi.org/10.1016/j.vetmic.2005.01.003
Okwumabua O, O’Connor M, Shull E (2003) A polymerase chain reaction (PCR) assay specific for Streptococcus suis based on the gene encoding the glutamate dehydrogenase. FEMS Microbiol Lett 218:79–84. https://doi.org/10.1111/j.1574-6968.2003.tb11501.x
Okura M, Maruyama F, Ota A, Tanaka T, Matoba Y, Osawa A, Sadaat SM, Osaki M, Toyoda A, Ogura Y, Hayashi T, Takamatsu D (2019) Genotypic diversity of Streptococcus suis and the S suis-like bacterium Streptococcus ruminantium in ruminants. Vet Res 50:94. https://doi.org/10.1186/s13567-019-0708-1
Gottschalk M, Lacouture S, Fecteau G, Desrochers A, Boa A, Saab ME, Okura M (2020) Isolation of Streptococcus ruminantium (Streptococcus suis-like) from diseased ruminants in Canada. Can Vet J 61:473–475
Neila-Ibáñez C, Pintado E, Velarde R, Aguilar FX, Vidal E, Aragon V, Abarca ML (2022) First report of Streptococcus Ruminantium in wildlife: phenotypic differences with a Spanish domestic ruminant isolate. Microbiol Res 13:102–113. https://doi.org/10.3390/microbiolres13010008
Dirksen G, Gründer HD, Stöber M (1993). Die klinische untersuchung des rindes. Begrundet von Rosenberger G. (third ed., Verlagbuchhandlung Paul Parey, Berlin and Hamburg.
Onni T, Vidili A, Bandino E, Marogna G, Schianchi G, Tola S (2012) Identification of coagulase-negative staphylococci isolated from caprine milk samples by PCR-RFLP of groEL gene. Small Rumin Res 104:185–190. https://doi.org/10.1016/j.smallrumres.2011.10.004
Rosa MN, Agnoletti F, Lollai S, Tola S (2019) Comparison of PCR-RFLP, API® 20 Strep and MALDI-TOF MS for identification of Streptococcus spp. collected from sheep and goat milk samples. Small Rumin Res 180:35–40. https://doi.org/10.1016/j.smallrumres.2019.09.023
CLSI (2023) Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals. 6th ed. CLSI supplement VET01S. Clinical and Laboratory Standards Institute.
EUCAST (2023). The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 13.0.
Vezina B, Rosa MN, Canu A, Tola S (2022) Genomic surveillance reveals antibiotic resistance gene transmission via phage recombinases within sheep mastitits-associated Streptococcus uberis. BMC Vet Res 18:264. https://doi.org/10.1186/s12917-022-03341-1
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Wick RR (2023) Dead-end count for QC of short-read assemblies (v1.0.0). Zenodo. https://doi.org/10.5281/zenodo.7662683
Nomoto R, Ishida-Kuroki K, Tohya M, Nakagawa I, Sekizaki T, Dunning Hotopp JC (2022) Complete genome sequences of three Streptococcus ruminantium strains obtained from endocarditis lesions of cattle in Japan. Microbiol Resour Announc 11:e01248-e1321. https://doi.org/10.1128/mra.01248-21
Tohya M, Sekizaki T, Miyoshi-Akiyama T (2018) Complete genome sequence of Streptococcus ruminantium sp. nov. GUT-187T (=DSM 104980T =JCM 31869T), the type strain of S. ruminantium, and comparison with genome sequences of Streptococcus suis strains. Genome Biol Evol 10:1180–1184. https://doi.org/10.1093/gbe/evy078
Kolmogorov M, Yuan J, Lin Y, Pevzner PA (2019) Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37:540–546. https://doi.org/10.1038/s41587-019-0072-8
Krueger F, James F, Ewels P, Afyounian E, Weinstein M, Schuster-Boeckler B, Hulselmans G (2023). FelixKrueger/TrimGalore: v0.6.10. Zenodo. https://doi.org/10.5281/zenodo.7598955
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arxiv. https://doi.org/10.4550/arXiv.1303.3997
Wick RR, Holt KE (2022) Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLoS Comput Biol 18:e1009802. https://doi.org/10.1371/journal.pcbi.1009802
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. https://doi.org/10.1038/s41467-018-07641-9
Harris SR (2018) SKA: Split Kmer analysis toolkit for bacterial genomic epidemiology. biorxiv. https://doi.org/10.1101/453142
Lees JA, Harris SR, Tonkin-Hill G, Gladstone RA, Lo SW, Weiser JN, Corander J, Bentley SD, Croucher NJ (2019) Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res 29:304–316. https://doi.org/10.1101/gr.241455.118
Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A (2021) Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 7:000685. https://doi.org/10.1099/mgen.0.000685
Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, Hoffmann M, Pettengill JB, Prasad AB, Tillman GE, Tyson GH, Klimke W (2021) AMRFinder plus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep 11:12728. https://doi.org/10.1038/s41598-021-91456-0
Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SDW, Corander J, Bentley SD, Parkhill J (2020) Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol 21:180. https://doi.org/10.1186/s13059-020-02090-4
Roodsant TJ, Van Der Putten BCL, Tamminga SM, Schultsz C, Van Der Ark KCH (2021) Identification of Streptococcus suis putative zoonotic virulence factors: a systematic review and genomic meta-analysis. Virulence 12:2787–2797. https://doi.org/10.1080/21505594.2021
Estrada AA, Gottschalk M, Rendahl A, Rossow S, Marshall-Lund L, Marthaler DG, Gebhart CJ (2021) Proposed virulence-associated genes of Streptococcus suis isolates from the United States serve as predictors of pathogenicity. Porcine Health Manag 7:22. https://doi.org/10.1186/s40813-021-00201-6
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinform 10:421. https://doi.org/10.1186/1471-2105-10-421
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
RStudio-Team (2020) RStudio: integrated development for R. RStudio, Boston
Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen TL, Miller E, Bache SM, Müller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H (2019) Welcome to the tidyverse. J Open Source Softw 4:1686. https://doi.org/10.2105/joss.01686
Neuwirth E (2014) RColorBrewer: ColorBrewer Palettes.
Csardi G, Nepusz T (2005) The Igraph software package for complex network research. Int J Complex Syst 1695.
Pedersen TL (2021) Package ‘ggforce’
Yu G (2020) aplot: Decorate a ‘ggplot’ with associated information.
Paradis EJC, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. https://doi.org/10.1093/bioinformatics/btg412
Yu G, Smith DK, Zhu H, Guan Y, Tsan-Yuk Lam T (2017) ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8:28–36. https://doi.org/10.1111/2041-210X.12628
Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K (2022) cluster: cluster analysis basics and extensions. R Package Version 2(1):4
Maron DF, Smith TJ, Nachman KE (2013) Restrictions on antimicrobial use in food animal production: an international regulatory and economic survey. Glob Health 9:48. https://doi.org/10.1186/1744-8603-9-48
Okwumabua O, Peterson H, Hsu HM, Bochsler P, Behr M (2017) Isolation and partial characterization of Streptococcus suis from clinical cases in cattle. J Vet Diagn Invest 29:160–168. https://doi.org/10.1177/1040638717690014
Okwumabua O, Williamson CHD, Pearson TR, Sahl JW (2020) Draft Genome Sequence of a Streptococcus suis isolate from a case of cattle meningitis. Microbiol Resour Announc 9:e00153-e220. https://doi.org/10.1128/MRA.00153-20
Sherwin VE, Green MJ, Leigh JA, Egan SA (2021) Assessment of the prevalence of Streptococcus uberis in dairy cow feces and implications for herd health. J Dairy Sci 104:12042–12052. https://doi.org/10.3168/jds.2021-20310
Acknowledgements
We are grateful to the farm vet, Dr Giuliana Sanna, for reporting the mastitis outbreak, to Dr Luca Bano (Istituto Zooprofilattico Sperimentale delle Venezie), Dr Alessandra Gazzola and Dr Antonio Marco Maisano (Istituto Zooprofilattico della Lombardia e Emilia Romagna) for kindly providing S. suis field isolates and SensititreTM ITISVE8 plates and for their valuable technical support.
Funding
This research was supported by funds from the Istituto Zooprofilattico Sperimentale della Sardegna.
Author information
Authors and Affiliations
Contributions
Conceptualization: ST. Methodology: ST. Formal analysis: BV, ST. Investigation: NMR, GM, AC and MM. Writing-Original Draft: BV, ST. Visualization: BV. Supervision: ST. Project administration: ST. Funding acquisition: ST. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Freddy Haesebrouck
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Streptococcus species included in the MBT Compass® Library Rev. K (2022).
Additional file 2:
Isolates used in this study and their relevant information including MIC and MIC breakpoints.
Additional file 3:
Protein fasta file of all putative virulence factors used in this analysis.
Additional file 4:
All R code used to generate results in this study
Additional file 5:
Genomic sequence of the gap gene and sequence similarity data for the S. ruminantium isolate n° 2622.
Additional file 6:
Restriction fragment length polymorphism (RFLP) patterns of PCR products from the gap gene of 12 S. ruminantium and 2 S. suis isolates after digestion with AluI enzyme and separated by 12% NuPAGE gel. Lanes 1-12, isolates from mastitis outbreak; c1, S. suis isolate 3089; c2, S. suis isolate 3627. M, Marker VIII (Roche).
Additional file 7:
Agarose electrophoresis of amplicons. Panel A, PCR products (688 bp) from the gdh gene of twelve (lanes 1-12) S. ruminantium and two S. suis isolates (c1=3089; c2=3627). Panel B, PCR products (336 bp) from the recN gene using the same isolates of panel A. Panel C, PCR products (240 bp) from 16S rRNA gene using the same isolates of panel A. M, Marker VIII (Roche).
Additional file 8:
SmaI-digested PFGE patterns of S. ruminantium isolated from 12 sheep with mastitis belonging to the same flock.
Additional file 9:
Output of pairwise SNV analysis as determined by SKA.
Additional file 10:
Histogram showing the distribution of pairwise SNVs between all S. ruminantium genomes used in this study.
Additional file 11:
Output of antimicrobial resistance analysis as determined by AMRFinder, presented in long table format.
Additional file 12
: Output of Twilight of the pangenome analysis, based on isolate source.
Additional file 13:
Summary of the putative virulence factors identified in two previous analyses from Streptococcus suis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rosa, M.N., Vezina, B., Marogna, G. et al. Streptococcus ruminantium-associated sheep mastitis outbreak detected in Italy is distinct from bovine isolates. Vet Res 54, 118 (2023). https://doi.org/10.1186/s13567-023-01248-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-023-01248-9