
Abstract
Duck enteritis virus (DEV) is a member of the Alphaherpesvirinae subfamily. The characteristics of some DEV genes have been reported. However, information regarding the DEV UL47 gene is limited. In this study, we identified the DEV UL47 gene encoding a late structural protein located in the nucleus of infected cells. We further found that two domains of DEV pUL47, amino acids (aa) 40 to 50 and 768 to 777, could function as nuclear localization sequence (NLS) to guide the nuclear localization of pUL47 and nuclear translocation of heterologous proteins, including enhanced green fluorescent protein (EGFP) and beta-galactosidase (β-Gal). Moreover, pUL47 significantly inhibited polyriboinosinic:polyribocytidylic acid [poly(I:C)]-induced interferon beta (IFN-β) production and downregulated interferon-stimulated gene (ISG) expression, such as Mx and oligoadenylate synthetase-like (OASL), by interacting with signal transducer and activator of transcription-1 (STAT1).
Similar content being viewed by others

Introduction
Duck virus enteritis (DVE), is an acute, septic, and febrile disease that occurs in waterfowl of all ages, with a high mortality rate and a declined duck egg production, resulting in severe economic losses to duck industry [1]. A variety of vaccines have been developed to control this disease [2]. DVE is caused by duck enteritis virus (DEV), which is a member of the Alphaherpesvirinae subfamily; the genome is a linear double-stranded DNA molecule (approximately 150 kb in length) divided into a unique long (UL) region and a unique short (US) region flanked by an internal short repeat (IRS) and a short terminal repeat (TRS) [3].
Several studies have been reported regarding the UL47 gene. The herpes simplex virus-1(HSV-1) UL47 gene encodes two major tegument proteins, VP13 and VP14, which represent differently processed forms of post-translational modification (phosphorylation, glycosylation or nucleotidylylation); VP13/14 is an RNA binding protein and shuttles between the nucleus and cytoplasm [4], modulates the trans-acting activity of trans-inducing factor VP16 and stimulates viral immediate-early gene transcription in newly infected cells [5]. In addition, VP13/14 can elicit T cell responses, and three epitopes of VP13/14, including the residues from 286 to 294, 504 to 512, and 544 to 552, were recognized by multifunctional CD8+ T cells [6, 7]. VP8, the homologue of VP13/14 in bovine herpesvirus 1 (BoHV-1), which is also phosphorylated and glycosylated, shuttles between the nucleus and cytoplasm and binds the mRNA of gB, gC and ICP0 [8,9,10]. Furthermore, VP8 interacts with STAT1, heat shock protein-60 (HSP60), and DNA damage response proteins to regulate IFN-β production, apoptosis, and mitochondrial functions, respectively [11,12,13]. Nevertheless, the infectious laryngotracheitis virus (ILTV) UL47 gene exhibits several differences from those of other alphaherpesviruses; the conserved UL47 gene is translocated from the UL to the US region and located between the conserved US3 and US4 genes [14]. The UL47 genes of HSV-1 [15], ILTV [14], pseudorabies virus (PRV) [16, 17], BoHV-1 [18], varicella-zoster virus (VZV) [19] and Marek’s disease virus (MDV-1) [20] have also been demonstrated to be dispensable for viral replication in cell culture.
The properties of some DEV genes have been preliminary characterized [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. We previously reported that DEV UL47-encoded protein (pUL47) modulates the distribution of UL41-encoded protein (pUL41) [37]. Together with the findings of previous studies [38, 39], we speculated that a close connection exists between pUL41 and pUL47, but the role of DEV pUL47 has not yet been defined. In this report, we explored the kinetics and location of pUL47, and we found DEV UL47 was a late gene encoding an abundant protein assembled in virion, and pUL47 was mainly localized to the nucleus in newly infected cells. In addition, residues 40 to 50 and 768 to 777 were necessary for nuclear localization of pUL47 and nuclear translocation of heterologous proteins. Moreover, we also explored and found pUL47 could interact with STAT1 to significantly inhibit IFN-β production and ISG expression.
Materials and methods
Viruses, cells and vectors
Duck embryo fibroblast (DEF) cells were cultured in minimal essential medium (MEM; Gibco, Meridian Road Rockford, USA) supplemented with 10% (v/v) foetal bovine serum (FBS; Gibco, Meridian Road Rockford, USA) at 37 °C with 5% CO2. In this study, the DEV CHv strain (GenBank: JQ647509.1) was isolated [40] and propagated on DEF cells with 2% FBS MEM. Virus-containing media was collected and centrifuged to remove large cellular debris, and then stored at − 80 °C until use.
The pcDNA3.1(+)-UL47, pcDNA3.1(+)-UL47Δ40-50, pcDNA3.1(+)-UL47Δ768-777, pEGFP-C2-SV40, pEGFP-C2-β-gal, pEGFP-C2-SV40-β-gal plasmid, and IFN-β-Luc reporter plasmid expressing firefly luciferase under the control of the duck IFN-β promoter were prepared in our laboratory [41].
Construction of the recombinant expression vector
All primers were designed by Primer Premier 5 software (Table 1). The wild-type DEV UL47 (GenBank: AFC61841.1) major antigenic domains (1417–2353 bp) were cloned into pMD18T (Novagen, Podenzano, Italy) and then sub-cloned into pET-32a (+) vector (Novagen, Podenzano, Italy) to create pET-32a (+)-UL47. We further constructed the eukaryotic plasmid pcDNA3.1(+)-Flag-UL47, and then deleted the 40 to 50 aa (RRSGKRRTLDR) and 768 to 777 aa (KALKRRLTGG) of pUL47, named pcDNA3.1(+)-Flag-UL47Δ40-50-768-777. Moreover, we fused residues 40 to 50 and 768 to 777 of DEV pUL47 to pEGFP-C2 or pEGFP-C2-β-gal, named pEGFP-C2-40-50, pEGFP-C2-768-777, pEGFP-C2-40-50-β-gal and pEGFP-C2-768-777-β-gal, respectively. In addition, the entire UL47 open reading frame (ORF) and duck STAT1 (Gene ID: 101795022) ORF were inserted into the pcaggs vector, named pcaggs-UL47-Flag and pcaggs-STAT1-myc, respectively. UL47 ORF with an LgBiT tag and STAT1 ORF with an SmBiT tag were inserted into the C- or N-termini of pcDNA3.1(+) vector, named pcDNA3.1-C-UL47-LgBiT and pcDNA3.1-N-STAT1-SmBiT, respectively.
Preparation of the polyclonal antibody
pET-32a(+)-UL47 was transformed into E. coli BL21 (DE3). Freshly transformed bacteria were inoculated into LB medium (100 µg/mL Amp) at 37 °C until the absorbance at 600 nm reached 0.4–0.6 and then expressed under the IPTG (0.2 mM) induction for 6 h at 37 °C. The vector control culture was analysed in parallel. The bacteria were collected by centrifugation (10 000 × g, 15 min, 4 °C) and disrupted by ultrasonication. The fusion protein was purified by gel and immunized in rabbit with Freund’s adjuvant to prepare a specific polyclonal antibody.
SDS-PAGE and western blotting analysis
Samples containing 20 µL of total proteins were boiled for 10 min; 5 µL of 5x SDS loading sample buffer was added, and the samples were centrifuged (10 000 × g, 5 min, 4 °C). Then, 10 µL of supernatant of each sample was analysed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). The samples were transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked for 2 h in 5% skim milk at 37 °C and then incubated with primary antibody and probed with HRP-conjugated secondary antibody (Bio-Rad Lab, CA, USA) for 1 h at 37 °C, respectively [43]. All antibodies were diluted in 1% skim milk. After several rinses with TBST (containing 0.1% Tween-20) to remove unbound antibodies, proteins were detected with Western BLoT Chemiluminescence HRP Substrate (Takara, Dalian, China) according to the manufacturer’s instructions. The following antibody were used: rabbit anti-DEV antibody (1:800), rabbit anti-UL47 polyclonal antibody (1:1000), mouse anti-actin antibody (1:5000, Proteintech-66009-1), mouse anti-GFP antibody (1:1000, Beyotime-AG281-1), mouse anti-Flag MAb (1:5000, MBL-M185-3) and mouse anti-Myc MAb (1:10 000, MBL-M192-3).
Indirect immunofluorescence assay (IFA)
DEV-infected DEF cells in 6-well dishes were collected on glass coverslips at 0, 9, 20, 36, 48, 60, and 72 h post-infection (hpi), fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.25% Triton X-100 for 30 min at 4 °C. The cells were rinsed three times with PBST (containing 0.1% Tween-20) for 5 min each time and blocked for 2 h with 5% BSA PBS at 37 °C. Then, the cells were incubated with primary antibodies and secondary antibodies. All antibodies were diluted in 1% BSA PBS. Finally, the cell nuclei were visualized with DAPI (Roche, Mannheim, Germany) for 15 min at room temperature. Coverslips were sealed with glycerine buffer and kept in a humidified chamber to avoid evaporation. The coverslips were visualized using a fluorescence microscope (Nikon ECLIPSE 80i, Japan).
RT-qPCR
Total RNA was collected using TRIzol reagent l (Invitrogen, CA, USA) according to the manufacturer’s recommendations, and complementary DNA (cDNA) was generated using the PrimeScript®RT reagent kit with the gDNAeraser (Takara, Beijing, China). Target genes were detected using the previously described primers (Table 1), and β-actin was used as internal reference as previously described [21]. All reactions were performed in triplicate and in at least three independent experiments. The relative levels of gene expression were determined with the 2−ΔΔCt method.
Luciferase reporter assays
DEF cells were co-transfected with 400 ng/well of pcaggs-UL47-Flag or an empty vector, 400 ng/well of reporter plasmid IFN-β-Luc, and 4 ng/well of pRL-TK (Promega, USA) using Lipofectamine 3000, and the cells were stimulated with 50 ng/ml poly(I:C) at 12 h after transfection and harvested at 24 h after stimulation. Finally, we detected the firefly luciferase activity by the dual-luciferase assay system (Promega, USA) according to the manufacturer’s recommendations. All reactions were performed in triplicate and at least three independent experiments.
NanoLuc Binary Technology (NanoBiT) assay [44]
BHK21 cells in growth medium were seeded in white 96-well plates (Costar 3917) at 37 °C and 5% CO2. The cells were co-transfected with pcDNA3.1-C-UL47-LgBiT and pcDNA3.1-N-STAT1-SmBiT using Lipofectamine 3000 according to the instructions; FKBP-SmBiT and FRB-LgBiT were used as positive controls, whereas Halotag-SmBiT coexpressed with pcDNA3.1-C-UL47-LgBiT, pcDNA3.1-C-UL47-LgBiT coexpressed with FKBP-SmBiT and pcDNA3.1-N-STAT1-SmBiT coexpressed with FRB-LgBiT were used as negative controls. The medium was replaced with serum-free media, and luciferase was detected after 20 h posttransfection. Luminescence was measured after addition of 25 μl Nano-Glo Live cell reagent (Promega, N2012) prepared according to the manufacturer’s protocol. All reactions were performed in triplicate and in at least three independent experiments.
Virion purification
DEV CHv strain was propagated in 9-day-old duck embryos. The embryos that died at 36–72 hpi were harvested. The allantoic fluids from the duck embryos were collected to purify the virion. In brief, clarified supernatants were underlaid with 5 mL 30% (w/v) sucrose in PBS and centrifuged in a Beckman Ti70 rotor (40 000 × g, 2 h, 4 °C). The pellet was finally diluted in PBS and stored at − 80 °C.
Mass spectrometry
Purified virion samples were boiled for 10 min with 5x SDS loading sample buffer and then analyzed by 12% SDS-PAGE. The whole gel was stained with Coomassie brilliant blue (Sigma) and then sent to Sangon Biotech Company (Shanghai, China) for liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis.
DEV UL47 expression kinetics
During the productive cycle, herpesviruses strictly exhibit temporal cascades of gene expression that are divided into three stages: immediate early (IE), early (E), and late (L); L genes strictly requires the onset or completion of lytic DNA amplification [36]. To identify the DEV UL47 expression kinetics, DEF cells were infected with DEV and incubated at 37 °C for 2 h with 300 µg/mL DNA polymerase inhibitor, ganciclovir (GCV) or 100 µg/mL protein synthesis inhibitor, cycloheximide (CHX), and the medium was replaced with 2% FBS MEM including the drugs. Total RNA was extracted at 24 hpi with TRIzol and subsequently reverse transcribed into cDNA. PCR was conducted using primers confirmed on a 1% agarose gel.
Statistical analysis
The comparisons between the different groups were made by one-way ANOVA with GraphPad Prism 7.0 software (La Jolla, CA, USA). All experiments were repeated at least three times individually. The data was expressed as the mean and standard error of the mean (SEM). Values of *p < 0.05 were considered statistically significant.
Results
Preparation of a specific rabbit anti-UL47 polyclonal antibody
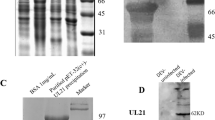
The expressed product was a UL47-his fusion protein (approximately 55 kDa) that was largely present in insoluble form but not in the vector control culture (Figure 1A). Next, the fusion protein was purified by gel and electric elusion (Figure 1B), detected with the rabbit anti-DEV antibody by western blotting [37] (Figure 1C), and then immunized in rabbit with Freund’s adjuvant to prepare a specific anti-UL47 polyclonal antibody. The pUL47 in the DEV-infected cells reacted strongly with this specific polyclonal antibody (Figure 2B).

Purified fusion protein and western blot analysis. A Lane 1, pET32a-UL47, non-induced; Lane 2, pET-32a-UL47 recombinant bacteria after induction in E. coli BL21 (DE3); Lane 3, pET-32a(+) vector; Lane 4, pET-32a-UL47 recombinant bacterial supernatant; Lane 5, pET-32a-UL47 recombinant bacterial sediment. B Purification of the recombinant protein. Lane 1, purified protein (approximately 55 kDa); C Expressed protein was recognized with rabbit anti-DEV antibody (1:800). Lane 1, recombinant protein (approximately 55 kDa); Lane 2, pET-32a (+) vector; M. Precision Plus Protein™ Dual Color Standards.

The DEV UL47 gene is a late gene. A Total RNA isolated from mock-infected and DEV-infected cells at various times was analysed using RT-qPCR and normalized to β-actin, and plotted the fold change over mock. B The expression analysis of the UL47 gene product in DEV-infected DEFs at different time points. The samples derived from the same experiment and gels were processed in parallel. C Schematic diagram showed the IE gene (UL54), E gene (UL13), and L gene (US2). D GCV group: DEV-infected cells treated with GCV; CHX group: DEV-infected cells treated with CHX. (+) group: DEV-infected cells without drugs. M, DL2000 Marker.
The DEV UL47 gene is a late gene
As shown in Figure 2A, the UL47 gene transcript level increased gradually from 6 hpi to 48 hpi and peaked at 48 hpi followed by a steady decrease.
We further identified the expression of the DEV UL47 gene. In lysates of DEV-infected cells, the UL47-specific antiserum recognized an 89 kDa major protein (Figure 2B). The band was not detected in the mock group and 6hpi and increased in amount until 48 hpi, with a steady decrease at 72 hpi. The β-actin antibody was used to assess β-actin as the loading control.
In addition, we treated the infected cells with GCV or CHX, and then collected them at 24 hpi to detect viral genes. The correct bands of β-actin (177 bp) and the IE gene UL54 (127 bp) [42], E gene UL13 (131 bp) [45], and L gene Us2 (111 bp) [36] were shown (Figure 2C), and the UL47 band (150 bp) was detected in the positive group but not in the GCV, CHX or negative groups (Figure 2D). These results showed that DEV UL47 gene was L gene due to its strict dependence on the early synthesis of viral DNA and protein.
pUL47 is a component of the virion
Mass spectrometry was used to identify the protein content of extracellular virions. The results showed that pUL47 was present in mature extracellular virions. Based on the exponentially modified protein abundance index (emPAI), the relative abundance of UL47 was far higher than that of gC, seventeen unique DEV UL47 peptides were detected, while three unique peptides matched the DEV gC (P < 0.05) (Table 2). Furthermore, the DEV pUL47 band was detected in virus-infected cells and virion particles (Figure 3A).

DEV pUL47 is a component of the virion. The purified DEV virions were identified using rabbit anti-UL47 antibody serum. As a control, no band was present in the DEF cell group.
DEV UL47 protein mainly localizes to the nucleus in newly infected cells
To analyse the intracellular localization of the pUL47, IFA was performed on DEF cells infected with DEV. The results showed that pUL47-specific fluorescence (green) was mainly located in the nucleus as well as diffused in the cytoplasm, and the signal became gradually stronger over time (Figure 4). In some nuclei, the protein accumulated in the nucleoli (9–36 hpi) and appeared diffuse (48–72 hpi). No fluorescence was observed in the mock-infected cells.

pUL47 mainly localizes in the nucleus of DEV-infected cells. Representative DEV pUL47 localization (green in all images). The mock group included untreated DEF cells using rabbit anti-UL47 polyclonal antibody and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 568 (Invitrogen, 1:1000) (magnification: 200×; scale bar: 10 μm).
Two small original motifs of UL47 function as an NLS
According to previous results [37], we further predicted the putative basic region of DEV pUL47 using software at https://www.predictprotein.org/. The corresponding sequences (residues 40 to 50 and 768 to 777 of pUL47) were fused to pEGFP-C2 and transfected into DEF cells. The fusion proteins EGFP-C2-40-50 and EGFP-C2-768-777 were mostly localized in the nucleus (Figure 5A). As controls, DEF cells were transfected with pEGFP-C2 or with a vector expressing EGFP carrying NLS of SV40 large T antigen, leading to cytoplasmic or nuclear localization, respectively.

The residues 40–50 and 768–777 of DEV pUL47 could direct heterologous proteins to the nucleus. All transfected samples were collected after 24 h posttransfection. A DEF cells were transfected with pEGFP-C2-SV40, pEGFP-C2-40-50, pEGFP-C2-768-777 and pEGFP-C2, respectively. B The expression of proteins was confirmed by Western blotting using mouse anti-GFP monoclonal antibody and goat anti-mouse IgG HRP-conjugated antibody (1:5000) (magnification: 200×; scale bar: 10 μm). C DEF cells were transfected with pEGFP-C2-SV40-β-gal, pEGFP-C2-40-50-β-gal, pEGFP-C2-768-777-β-gal and pEGFP-C2-β-gal, respectively. D The expression of proteins was confirmed by western blotting using mouse anti-GFP monoclonal antibody.
Because of the small molecular weight of EGFP, which enables its passive diffusion, we further constructed the plasmids pEGFP-C2-40-50-β-gal and pEGFP-C2-768-777-β-gal, encoding a fusion protein containing a large-galactosidase cytoplasmic protein, avoiding the passive diffusion phenomenon. As shown in Figure 5B, the fusion proteins EGFP-C2-40-50-β-gal and EGFP-C2-768-777-β-gal were distributed in the nuclei of DEF cells. As controls, the DEF cells were transfected pEGFP-C2-β-gal and pEGFP-C2-SV40-β-gal showed an exclusively cytoplasmic and nuclear localization, respectively. The following results showed that these two small original motifs should harbour the main NLS.
The amino acids 40 to 50 and 768 to 777 of DEV pUL47 are necessary for nuclear localization
According to the Ref. [37], we found that the residues 40 to 50 and 768 to 777 affected the nuclear localization of pUL47. In addition, we further identified that these two signals could translocate the EGFP and β-gal into the nucleus. When the residues 40 to 50 and 768 to 777 of pUL47 were deleted, the mutated protein (green) was mostly located in the cytoplasm, a small part of the pUL47 was still located in the nucleus (Figure 6A), which showed that DEV pUL47 might have another fragment to affect nuclear localization.

The residues 40 to 50 and 768 to 777 of DEV pUL47 are necessary for nuclear localization. A Distribution of the UL47 gene in DEF cells transfected with pcDNA3.1-Flag-UL47Δ40–50-768-777 (magnification: 200×; scale bar: 10 μm). B The expression of proteins was confirmed by western blotting using rabbit anti-UL47 polyclonal antibody.
DEV pUL47 inhibits IFN-β signalling by targeting STAT1
We further explored whether the pUL47 could inhibit IFN-β signalling. DEF cells were co-transfected with UL47-Flag plasmid or an empty vector and IFN-β-luc reporter plasmid, stimulated by the dsRNA analogues poly(I:C) stimulator, and then subjected to dual-luciferase reporter (DLR) assays. As shown in Figure 7A, DEV pUL47 significantly inhibited the IFN-β-luc activity induced by poly (I:C).

DEV UL47 inhibits IFN-β signalling induced by poly(I:C). All transfected samples were collected at 36 h posttransfection. A DEV UL47 inhibited the IFN-β-luc luciferase activation induced by poly(I:C) in DEF cells. DEF cells were co-transfected with IFN-β-luc luciferase reporter plasmid, pRL-TK, pcaggs-UL47-Flag or an empty vector, and then stimulated by 50 µg/mL at 12 h posttransfection. The cells were harvested and detected by dual-luciferase assay at 24 h posttransfection. Protein expression was confirmed by western blotting using mouse anti-Flag MAb. All experiments were performed in triplicate (n = 3). The data was analyzed by one-way ANOVA. ***p < 0.001
According to previous reports, BoHV-1 VP8 inhibited IFN-β signalling by interacting with and preventing the nuclear localization of STAT1 [11]. Therefore, we explored the relationship between DEV pUL47 and duck STAT1. DEV pUL47 co-localized with STAT1 in the cytoplasm (Figure 8A). Next, we detected the interaction between UL47 and STAT1, and the experimental group (co-transfected with pcDNA3.1-C-UL47-LgBiT and pcDNA3.1-N-STAT1-SmBiT) demonstrated a stronger signal than the positive group (co-transfected with FKBP-SmBiT and FRB-LgBiT); however, the negative control groups were not activated (Figure 8B). These results showed that DEV pUL47 interacted with STAT1 in the cytoplasm. Based on these results, we further explored whether pUL47 inhibited the STAT1-induced ISG expression. DEF cells were co-transfected with UL47-Flag or empty vector with STAT1-myc and then detected by RT-qPCR. As shown in Figure 8C, DEV pUL47 significantly reduced the expression of Mx and OASL proteins induced by STAT1. These results showed that DEV pUL47 targeted STAT1 to inhibit IFN-β signalling and host innate immunity.

pUL47 interacted with STAT1 in the cytoplasm and inhibited ISG expression induced by STAT1. A HEK293T cells were coexpressed pcaggs-STAT1-myc with pcaggs-UL47-Flag, pcaggs-STAT1-myc with an empty vector and pcaggs-UL47-Flag with an empty vector (magnification: 200×; scale bar: 10 μm). B BHK21 cells contained pcDNA3.1-C-UL47-LgBiT coexpressed with pcDNA3.1-N-STAT1-SmBiT, FKBP-SmBiT coexpressed with FRB-LgBiT as positive controls, Halotag-SmBiT coexpressed with pcDNA3.1-C-UL47-LgBiT, pcDNA3.1-C-UL47-LgBiT coexpressed with FKBP-SmBiT and pcDNA3.1-N-STAT1-SmBiT coexpressed with FRB-LgBiT as negative controls. The luciferase signal was detected via NanoBiT after 20-h posttransfection. C DEV UL47 inhibited the mRNA levels of Mx and OASL induced by STAT1. DEF cells were co-transfected with pcaggs-STAT1-myc and pcaggs-UL47-Flag or an empty vector. The cells were harvested and detected by RT-qPCR at 36 h posttransfection. Protein expression was confirmed by Western blotting. All experiments were performed in triplicate (n = 3). The data was analyzed by one-way ANOVA. *P < 0.05, **P < 0.01 and ****P < 0.0001.
Discussion
Herpesviruses assemble more than 30 proteins into mature virions, which contain the nucleoprotein core, capsid shell, tegument and envelope. The tegument, which is a complex structure contained numerous viral gene products that play crucial roles in viral replication and the interaction of the virus with the host immune system [46, 47]. The UL47 gene is conserved in the Alphaherpesvirinae subfamily [48, 49], and pUL47 as a late abundant tegument protein in HSV-1 [50], BoHV-1 [51], MDV [52], ILTV [53] and PRV [54]. In addition, HSV-1 VP13/14 forms a complex with the viral proteins UL34, UL31, and/or US3, which regulate primary envelopment during viral nuclear egress [55]. Further research showed that VP13/14 interacts with the capsid protein pUL17, providing another link between the tegument and nucleocapsid [56]. PRV pUL47 plays a role in virion morphogenesis at the late stage of infection, and deletion of UL47 impairs the secondary envelopment, resulting in intracytoplasmic aggregation of tegumented capsids [16]. MDV pUL47 also regulates the expression of the UL46-UL49 locus [52]. However, BoHV-1 UL47 cannot affect the secondary envelopment [18]. In this study, we further found that the DEV UL47 gene encoded a late abundant structural protein assembled in the virion. Based on the high level of emPAI, the large abundance of this protein indicates an important function, requiring further exploration of the role of DEV pUL47 in the viral life cycle.
All the UL47 proteins encoded by HSV-1 [57], BoHV-1 [10], ILTV [53] and PRV [16] exhibit nuclear localization, suggesting an important role of these proteins in the nucleus. In addition, VP13/14 and VP8 can bind RNA and play a pivotal role in RNA transport. Our results have also shown that DEV pUL47 is mainly localized in the nucleus at early stage of infection and diffused in the cytoplasm at the late stage of infection. We speculated that DEV pUL47 could shuttle between the nucleus and cytoplasm. Whether DEV pUL47 has the same function to modulate the transcription remains to be elucidated.
Nuclear localization mainly depends on the nuclear targeting signal. The best-characterized nuclear targeting signal is the classical nuclear localization sequence (cNLS) [58, 59]. The cNLS are formed by one or two basic clusters of amino acid residues, termed monopartite or bipartite NLSs, respectively, and have the following consensus sequences: K(K/R)X(K/R) for monopartite cNLS and KRX10-12K(K/R)X(K/R) for bipartite cNLS (X corresponds to any residue) [60]. We predicted two putative basic regions of DEV pUL47 using software: monopartite NLS in the N-terminus (40RRSGKRRTLDR50) and C-terminus (768KALKRRLTGG777). In this report, we further identified that these sequences affect the UL47 distribution and direct EGFP and β-gal proteins into the nucleus. However, no cNLS region exists in VP13/14 or VP8; others have identified nonclassical arginine-rich nuclear localization signals at the N-terminus of VP13/14 and VP8, which were sufficient to direct a heterologous protein to the nucleus [4, 10], and the arginine-rich NLS of VP13/14 is associated with the RNA binding motif [61]. We also found a similar arginine-rich region in DEV pUL47 (125RRRR128); however, the prediction outcomes showed that the NLS score is negative. Based on the prediction outcomes, we deleted the residues 40 to 50 and 768 to 777 of DEV pUL47, a small part of pUL47 was still located in the nucleus (Figure 6A). Therefore, we speculated that the motif may also affect the nuclear localization.
In addition, VP13/14 forms a stable complex with viral kinase US3 and they reciprocally regulate subcellular localization in infected cells; US3 phosphorylation of VP13/14 (Ser-77) near its NLS (residues 50 to 68) appears to promote nuclear localization of VP13/14 in infected cells [62]. Furthermore, VP8 is also phosphorylated by US3, and the phosphorylation sites are near putative NLSs of VP8 [63]. The phosphorylation site of a protein near its NLS mediates its nuclear localization for a number of viral proteins [64]. Therefore, in addition to specific signals, we speculated that DEV pUL47 as a physiological phosphorylation substrate of cellular or viral protein kinase to mediate its nuclear localization. Therefore, whether DEV pUL47 is modified by other protein kinase need further investigation.
The host innate immune responses are triggered by recognition of pathogen-associated molecular patterns (PAMPs) via host–pathogen recognition receptors (PRRs), leading to induction of IFNs and pro-inflammatory cytokines. IFNs then activate the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signalling pathway, resulting in transcription of numerous IFN-stimulated genes (ISGs). However, viruses can establish an infection in the host cells by overcoming the various innate defence mechanisms [65]. HSV-2 ICP27 directly associates with IRF3 [66], and US11 targets TBK1 to evade the antiviral innate immune response [67]. DEV is immunosuppressive and persistently infects monocytes/macrophages [68], but the mechanism of immune evasion by DEV is unknown. In our study, we first found that DEV pUL47 significantly inhibits the IFN-β signalling induced by poly(I:C) and downregulates the STAT1-induced ISG expression. Moreover, we also found that DEV pUL47 interacts with STAT1, similar to VP8 [11], but the detailed mechanism requires further exploration. Our data provide new insights into viral protein antagonizing host innate immune response in DEV infection.
In conclusion, the DEV UL47 gene is a late gene encoding an abundant structural protein assembled in virion. DEV pUL47 is mainly distributed in the nucleus of infected cells and contains two NLS motifs (residues 40 to 50 and 768 to 777). Moreover, DEV pUL47 inhibits IFN-β signalling by targeting STAT1 in the cytoplasm.
Availability of data and materials
The datasets analysed in this study are available from the corresponding author upon reasonable request.
Abbreviations
- β-Gal:
-
beta-galactosidase
- BoHV-1:
-
bovine herpesvirus 1
- DEV:
-
duck enteritis virus
- DP:
-
duck plague
- EGFP:
-
enhanced green fluorescent protein
- emPAI:
-
exponentially modified protein abundance index
- HSP60:
-
heat shock protein-60
- HSV-1:
-
herpes simplex virus-1
- ILTV:
-
infectious laryngotracheitis virus
- IFN:
-
interferon
- ISG:
-
interferon-stimulated gene
- MDV:
-
Marek’s disease virus
- NanoBiT:
-
nanoLuc Binary Technology
- NLS:
-
nuclear localization sequence
- NPCs:
-
nuclear pore complexes
- OASL:
-
oligoadenylate synthetase-like
- PAMPs:
-
pathogen-associated molecular patterns
- PRRs:
-
pathogen recognition receptors
- poly(I:C):
-
polyriboinosinic:polyribocytidylic acid
- PRV:
-
pseudorabies virus
- RT-qPCR:
-
real-time quantitative reverse-transcription PCR
- STAT1:
-
signal transducer and activator of transcription-1
- VZV:
-
varicella-zoster virus
References
Dhama K, Kumar N, Saminathan M, Tiwari R, Karthik K, Kumar M, Palanivelu M, Shabbir M, Malik Y, Singh R (2017) Duck virus enteritis (duck plague)-a comprehensive update. Vet Q 37(1):57–80
Huang J, Jia R, Wang M, Shu B, Yu X, Zhu D, Chen S, Yin Z, Chen X, Cheng A (2014) An attenuated Duck Plague Virus (DPV) vaccine induces both systemic and mucosal immune responses to protect Ducks against virulent DPV infection. Clin Vacc Immunol Cvi 21(4):457–462
Wu Y, Cheng A, Wang M, Yang Q, Zhu D, Jia R, Chen S, Zhou Y, Wang X, Chen X (2012) Complete genomic sequence of Chinese virulent duck enteritis virus. J Virol 86(10):5965
Donnelly M, Elliott G (2001) Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J Virol 75(6):2566–2574
Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79(15):9566–9571. https://doi.org/10.1128/JVI.79.15.9566-9571.2005
Srivastava R, Khan AA, Garg S, Syed SA, Furness JN, Vahed H, Pham T, Yu HT, Nesburn AB, BenMohamed L (2017) Human asymptomatic epitopes identified from the herpes simplex virus tegument protein VP13/14 (UL47) preferentially recall polyfunctional effector memory CD44 high CD62L low CD8 + TEM cells and protect humanized HLA-A*02:01 transgenic mice against ocular herpesvirus infection. J Virol 91(2):e01793. https://doi.org/10.1128/JVI.01793-16
Platt RJ, Khodai T, Townend TJ, Bright HH, Cockle P, Perez-Tosar L, Webster R, Champion B, Hickling TP, Mirza F (2013) CD8 + T lymphocyte epitopes from the Herpes simplex virus type 2 ICP27, VP22 and VP13/14 proteins to facilitate vaccine design and characterization. Cells 2(1):19–42. https://doi.org/10.3390/cells2010019
Labiuk SL, Babiuk LA, van Drunen Littel-van den Hurk S (2009) Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J Gen Virol 90(Pt 12):2829–2839. https://doi.org/10.1099/vir.0.013532-0
Islam A, Schulz S, Afroz S, Babiuk LA, van Drunen Littel-van den Hurk S (2015) Interaction of VP8 with mRNAs of bovine herpesvirus-1. Virus Res 197:116–126. https://doi.org/10.1016/j.virusres.2014.12.017
Zheng C, Brownlie R, Babiuk LA (2004) Characterization of nuclear localization and export signals of the major tegument protein VP8 of bovine herpesvirus-1 ☆. Virology 324(2):327–339
Afroz S, Brownlie R, Fodje M, van Drunen Littel-van den Hurk S (2016) VP8, the major tegument protein of Bovine Herpesvirus 1, interacts with cellular STAT1 and inhibits interferon beta signaling. J Virol 90(10):4889–4904. https://doi.org/10.1128/JVI.00017-16
Afroz S, Garg R, Fodje M, van Drunen Littel-van den Hurk S (2018) The major tegument protein of Bovine Herpesvirus 1, VP8, interacts with DNA damage response proteins and induces apoptosis. J Virol 92(15):e00773
Afroz S, Brownlie R, Fodje M, van Drunen Littel-van den Hurk S (2019) The bovine herpesvirus-1 major tegument protein, VP8, interacts with host HSP60 concomitant with deregulation of mitochondrial function. Virus Res 261:37–49. https://doi.org/10.1016/j.virusres.2018.12.006
Helferich D, Veits J, Teifke JP, Mettenleiter TC, Fuchs W (2007) The UL47 gene of avian infectious laryngotracheitis virus is not essential for in vitro replication but is relevant for virulence in chickens. J Gen Virol 88(3):732–742. https://doi.org/10.1099/vir.0.82533-0
Roizman B (2001) Herpes simplex viruses and their replication. Virology 19(2):2231–2295
Kopp M, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC (2002) Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J Virol 76(17):8820–8833. https://doi.org/10.1128/jvi.76.17.8820-8833.2002
Fuchs W, Granzow H, Mettenleiter TC (2003) A pseudorabies virus recombinant simultaneously lacking the major tegument proteins encoded by the UL46, UL47, UL48, and UL49 genes is viable in cultured cells. J Virol 77(23):12891–12900. https://doi.org/10.1128/jvi.77.23.12891-12900.2003
Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA, van Drunen Littel-van den Hurk S (2010) A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol 84(1):445–458. https://doi.org/10.1128/JVI.01544-09
Che X, Reichelt M, Sommer MH, Rajamani J, Zerboni L, Arvin AM (2008) Functions of the ORF9-to-ORF12 gene cluster in varicella-zoster virus replication and in the pathogenesis of skin infection. J Virol 82(12):5825–5834. https://doi.org/10.1128/JVI.00303-08
Dorange F, Tischer BK, Vautherot JF, Osterrieder N (2002) Characterization of Marek’s Disease Virus Serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J Virol 76(4):1959–1970
Zhang D, Lai M, Cheng A, Wang M, Wu Y, Yang Q, Liu M, Zhu D, Jia R, Chen S (2017) Molecular characterization of the duck enteritis virus US10 protein. Virol J 14(1):183
Jing Y, Ying W, Sun K, Wang M, Cheng A, Chen S, Jia R, Zhu D, Liu M, Yang Q (2017) Role of duck plague virus glycoprotein C in viral adsorption: absence of specific interactions with cell surface heparan sulfate. J Integrat Agric 16(5):1145–1152
Liu C, Cheng A, Wang M, Chen S, Jia R, Zhu D, Liu M, Sun K, Yang Q, Chen X (2016) Characterization of nucleocytoplasmic shuttling and intracellular localization signals in Duck Enteritis Virus UL54. Biochimie 127:86–94
Wu Y, Cheng A, Wang M, Zhang S, Zhu D, Jia R, Luo Q, Chen Z, Chen X (2011) Serologie detection of duck enteritis virus using an indirect ELISA based on recombinant UL55 protein. Avian Dis 55(4):626–632
Dai B, Cheng A, Wang M (2013) Characteristics and functional roles of VP5 protein of herpesviruses. Rev Med Microbiol 24(2):35–40
Wang M, Lin D, Zhang S, Zhu D, Jia R, Chen X, Cheng A (2011) Prokaryotic expression of the truncated duck enteritis virus UL27 gene and characteristics of UL27 gene and its truncated product. Acta Virol 56(4):323–328
Cheng A, Zhang S, Zhang X, Wang M, Zhu D, Jia R, Chen X (2012) Prokaryotic expression and characteristics of duck enteritis virus UL29 gene. Acta Virol 56(4):293
Zhang S, Xiang J, Cheng A, Wang M, Wu Y, Yang X, Zhu D, Jia R, Luo Q, Chen Z (2011) Characterization of duck enteritis virus UL53 gene and glycoprotein K. Virol J 8(1):1–9
Li L, Cheng A, Wang M, Xiang J, Yang X, Zhang S, Zhu D, Jia R, Luo Q, Zhou Y (2011) Expression and characterization of duck enteritis virus gI gene. Virol J 8(1):241
Wei X, Cheng A, Wang M, Hua C, Zhu D, Luo Q, Jia R, Chen X (2009) Expression and characterization of the UL31 protein from duck enteritis virus. Virol J 6(1):19
Lu L, Cheng A, Wang M, Jiang J, Zhu D, Jia R, Luo Q, Liu F, Chen Z, Chen X (2010) Polyclonal antibody against the DPV UL46M protein can be a diagnostic candidate. Virol J 7(1):1–10
Cai M, Cheng A, Wang M, Zhao L, Zhu D, Luo Q, Liu F, Chen X (2009) His6-tagged UL35 protein of duck plague virus: expression, purification, and production of polyclonal antibody. Intervirology 52(3):141–151
Shen C, Cheng A, Wang M, Sun K, Jia R, Tao S, Na Z, Zhu D, Luo Q, Yi Z (2010) Development and evaluation of an immunochromatographic strip test based on the recombinant UL51 protein for detecting antibody against duck enteritis virus. Virol J 7(1):268
Xiang J, Ma G, Zhang S, Cheng A, Wang M, Zhu D, Jia R, Luo Q, Chen Z, Chen X (2010) Expression and intracellular localization of duck enteritis virus pUL38 protein. Virol J 7(1):162
Xin H, Cheng A, Wang M, Jia R, Shen C, Chang H (2009) Identification and characterization of a duck enteritis virus US3-like gene. Avian Dis 53(3):363–369
Gao J, Cheng A, Wang M, Jia R, Zhu D, Chen S, Liu M, Liu F, Yang Q, Sun KF (2015) Identification and characterization of the duck enteritis virus (DEV) US2 gene. Genet Mol Res Gmr 14(4):13779
He T, Wang M, Cao X, Cheng A, Wu Y, Yang Q, Liu M, Zhu D, Jia R, Chen S (2018) Molecular characterization of duck enteritis virus UL41 protein. Virol J 15(1):12
Shu M, Taddeo B, Roizman B (2013) The nuclear-cytoplasmic shuttling of virion host shutoff RNase is enabled by pUL47 and an embedded nuclear export signal and defines the sites of degradation of AU-rich and stable cellular mRNAs. J Virol 87(24):13569–13578. https://doi.org/10.1128/jvi.02603-13
Shu M, Taddeo B, Zhang W, Roizman B (2013) Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc Natl Acad Sci USA 110(18):E1669–E1675. https://doi.org/10.1073/pnas.1305475110
Wu Y, Cheng A, Wang M, Zhu D, Jia R, Chen S, Zhou Y, Chen X (2012) Comparative genomic analysis of duck enteritis virus strains. J Virol 86(24):13841–13842. https://doi.org/10.1128/JVI.01517-12
Chen S, Liu P, He Y, Yang C, Wang M, Jia R, Zhu D, Liu M, Yang Q, Wu Y, Cheng A (2018) The 164 K, 165 K and 167 K residues in 160YPVVKKPKLTEE171 are required for the nuclear import of goose parvovirus VP1. Virology 519:17–22. https://doi.org/10.1016/j.virol.2018.03.020
Liu C, Cheng A, Wang M, Chen S, Jia R, Zhu D, Liu M, Sun K, Yang Q, Chen X (2015) Duck enteritis virus UL54 is an IE protein primarily located in the nucleus. Virol J 12(1):198
Chang H, Cheng A, Wang M, Zhu D, Jia R, Liu F, Chen Z, Luo Q, Chen X, Zhou Y (2010) Cloning, expression and characterization of gE protein of duck plague virus. Virol J 7:120. https://doi.org/10.1186/1743-422X-7-120
Zhang W, Jiang B, Zeng M, Duan Y, Wu Z, Wu Y, Wang T, Wang M, Jia R, Zhu D, Liu M, Zhao X, Yang Q, Wu Y, Zhang S, Liu Y, Zhang L, Yu Y, Pan L, Chen S, Cheng A (2020) Binding of Duck Tembusu virus nonstructural protein 2A to duSTING disrupts the induction of its signal transduction cascade to inhibit IFN-β induction. J Virol 94(9):e01850
Hu X, Wang M, Chen S, Jia R, Zhu D, Liu M, Yang Q, Sun K, Chen X, Cheng A (2017) The duck enteritis virus early protein, UL13, found in both nucleus and cytoplasm, influences viral replication in cell culture. Poult Sci 96(8):2899–2907
Crump C (2018) Virus Assembly and Egress of HSV. Adv Exp Med Biol 1045:23–44. https://doi.org/10.1007/978-981-10-7230-7_2
Guo H, Shen S, Wang L, Deng H (2010) Role of tegument proteins in herpesvirus assembly and egress. Protein Cell 1(11):987–998. https://doi.org/10.1007/s13238-010-0120-0
Mettenleiter TC (2002) Herpesvirus assembly and egress. J Virol 76(4):1537–1547. https://doi.org/10.1128/jvi.76.4.1537-1547.2002
Fuchs W, Granzow H, Klupp BG, Karger A, Michael K, Maresch C, Klopfleisch R, Mettenleiter TC (2007) Relevance of the interaction between alphaherpesvirus UL3.5 and UL48 proteins for virion maturation and neuroinvasion. J Virol 81(17):9307–9318. https://doi.org/10.1128/jvi.00900-07
Loret S, Guay G, Lippe R (2008) Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82(17):8605–8618. https://doi.org/10.1128/JVI.00904-08
Robinson KE, Meers J, Gravel JL, McCarthy FM, Mahony TJ (2008) The essential and non-essential genes of Bovine herpesvirus 1. J Gen Virol 89(Pt 11):2851–2863. https://doi.org/10.1099/vir.0.2008/002501-0
Jarosinski KW, Vautherot JF (2015) Differential expression of Marek’s disease virus (MDV) late proteins during in vitro and in situ replication: role for pUL47 in regulation of the MDV UL46-UL49 gene locus. Virology 484:213–226. https://doi.org/10.1016/j.virol.2015.06.012
Helferich D, Veits J, Mettenleiter TC, Fuchs W (2007) Identification of transcripts and protein products of the UL31, UL37, UL46, UL47, UL48, UL49 and US4 gene homologues of avian infectious laryngotracheitis virus. J Gen Virol 88(Pt 3):719–731. https://doi.org/10.1099/vir.0.82532-0
Kramer T, Greco TM, Enquist LW, Cristea IM (2011) Proteomic characterization of pseudorabies virus extracellular virions. J Virol 85(13):6427–6441. https://doi.org/10.1128/JVI.02253-10
Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y (2014) Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88(9):4657–4667. https://doi.org/10.1128/JVI.00137-14
Scholtes LD, Yang K, Li LX, Baines JD (2010) The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J Virol 84(15):7642–7650. https://doi.org/10.1128/JVI.00277-10
Yedowitz JC, Kotsakis A, Schlegel EFM, Blaho JA (2005) Nuclear localizations of the herpes simplex virus type 1 tegument proteins VP13/14, vhs, and VP16 precede VP22-dependent microtubule reorganization and VP22 nuclear import. J Virol 79(8):4730–4743. https://doi.org/10.1128/jvi.79.8.4730-4743.2005
Marelli M, Dilworth DJ, Wozniak RW, Aitchison JD (2001) The dynamics of karyopherin-mediated nuclear transport. Biochem Cell Biol. 79(5):603–612
Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NF, Curmi PM, Forwood JK, Boden M (1813) Kobe B (2011) Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta 9:1562–1577. https://doi.org/10.1016/j.bbamcr.2010.10.013
Macara IG (2001) Transport into and out of the Nucleus. Microbiol Mol Biol Rev 65(4):570–594
Donnelly M, Verhagen J, Elliott G (2007) RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J Virol 81(5):2283–2296. https://doi.org/10.1128/JVI.01677-06
Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y (2011) Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85(18):9599
Verhagen J, Hutchinson I, Elliott G (2006) Nucleocytoplasmic shuttling of bovine herpesvirus 1 UL47 protein in infected cells. J Virol 80(2):1059–1063. https://doi.org/10.1128/JVI.80.2.1059-1063.2006
Shen W, Westgard E, Huang L, Ward MD, Osborn JL, Chau NH, Collins L, Marcum B, Koach MA, Bibbs J (2008) Nuclear trafficking of the human cytomegalovirus pp71 (ppUL82) tegument protein. Virology 376(1):42–52
Tognarelli EI, Palomino TF, Corrales N, Bueno SM, Kalergis AM, Gonzalez PA (2019) Herpes simplex virus evasion of early host antiviral responses. Front Cell Infect Microbiol 9:127. https://doi.org/10.3389/fcimb.2019.00127
Guan X, Zhang M, Fu M, Luo S, Hu Q (2019) Herpes simplex virus type 2 immediate early protein ICP27 inhibits IFN-beta production in mucosal epithelial cells by antagonizing IRF3 activation. Front Immunol 10:290. https://doi.org/10.3389/fimmu.2019.00290
Liu X, Main D, Ma Y, He B (2018) Herpes simplex virus 1 inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J Virol 92(14):e00402–e00418. https://doi.org/10.1128/JVI.00402-18
Tian B, Cai D, He T, Deng L, Wu L, Wang M, Jia R, Zhu D, Liu M, Yang Q, Wu Y, Zhao X, Chen S, Zhang S, Huang J, Ou X, Mao S, Yu Y, Zhang L, Liu Y, Cheng A (2020) Isolation and selection of duck primary cells as pathogenic and innate immunologic cell models for duck plague virus. Front Immunol. https://doi.org/10.3389/fimmu.2019.03131
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Key Research and Development Program of China (2017YFD0500800), China Agricultural Research System (CARS-42-17), Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (SCCXTD-2020-18) and Integration and Demonstration of Key Technologies for Goose Industrial Chain in Sichuan Province (2018NZ0005).
Author information
Authors and Affiliations
Contributions
TH carried out the experiments and drafted the manuscript. AC and MW critically revised the manuscript and experimental design. QY, RJ, YW, HJ, SC, XZ, ML, DZ, SZ, XO, SM, QG, DS, XW, BT, YL, YY, LZ, LP and XC helped with the experiments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Sichuan Agricultural University (2016–17). Experiments were conducted in accordance with approved guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, T., Wang, M., Cheng, A. et al. Duck enteritis virus pUL47, as a late structural protein localized in the nucleus, mainly depends on residues 40 to 50 and 768 to 777 and inhibits IFN-β signalling by interacting with STAT1. Vet Res 51, 135 (2020). https://doi.org/10.1186/s13567-020-00859-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-020-00859-w